



Shreya Chenni, Freiberuflicher Verfasser von Rechtsvorschriften for medical devices, provides a 10-minute guide to FDA design controls for your Medizinprodukt.
Eine der Hauptursachen für Rückrufe von Medizinprodukten ist das Fehlen von Konstruktionskontrollen, wie von der FDA festgestellt [3,3a]. Daraufhin wurden die GMP-Vorschriften für Medizinprodukte um die Vorproduktionskontrollen ergänzt. Konstruktionskontrollen sind eine Reihe miteinander verbundener Praktiken und Verfahren, die in den Konstruktions- und Entwicklungsprozess integriert sind, d. h. ein System von Kontrollen und Gegenkontrollen. Auslegungskontrollen erhöhen die Wahrscheinlichkeit, dass die auf die Produktion übertragene Auslegung in ein Produkt umgesetzt wird, das für die vorgesehene Verwendung geeignet ist.
Anwendbarkeit
Alle Produkte der Klassen II und III sowie die folgenden Produkte der Klasse I werden einer Auslegungskontrolle unterzogen:
- Devices automated with computer software
- 868.6810 Katheter, tracheobronchiale Absaugung
- 878.4460 Handschuhe, Chirurgenhandschuhe
- 880.6760 Rückhalteeinrichtungen, Schutzvorrichtungen
- 892.5650 System, Applikator, Radionuklid, manuell
- 892.5740 Strahlenquelle, Radionuklid-Teletherapie
- Mit Computersoftware automatisierte Geräte
- Tracheobronchiale Absaugkatheter
- Chirurgenhandschuhe
- Schützende Fesseln
- System, Radionuklid, Applikator, manuell
- Quelle, Radionuklid-Teletherapie
Design controls apply to all D&D activities – for novel or improved devices being developed in the pre-market phase, as well as for changes to existing, marketed devices. Design controls do not apply to Forschung activities conducted during the proof of concept stage.
Design controls can be applied to any Produktentwicklung process. The following flowcharts are examples of the design controls applied to a traditional Waterfall design process and V-model process for Software (SW).
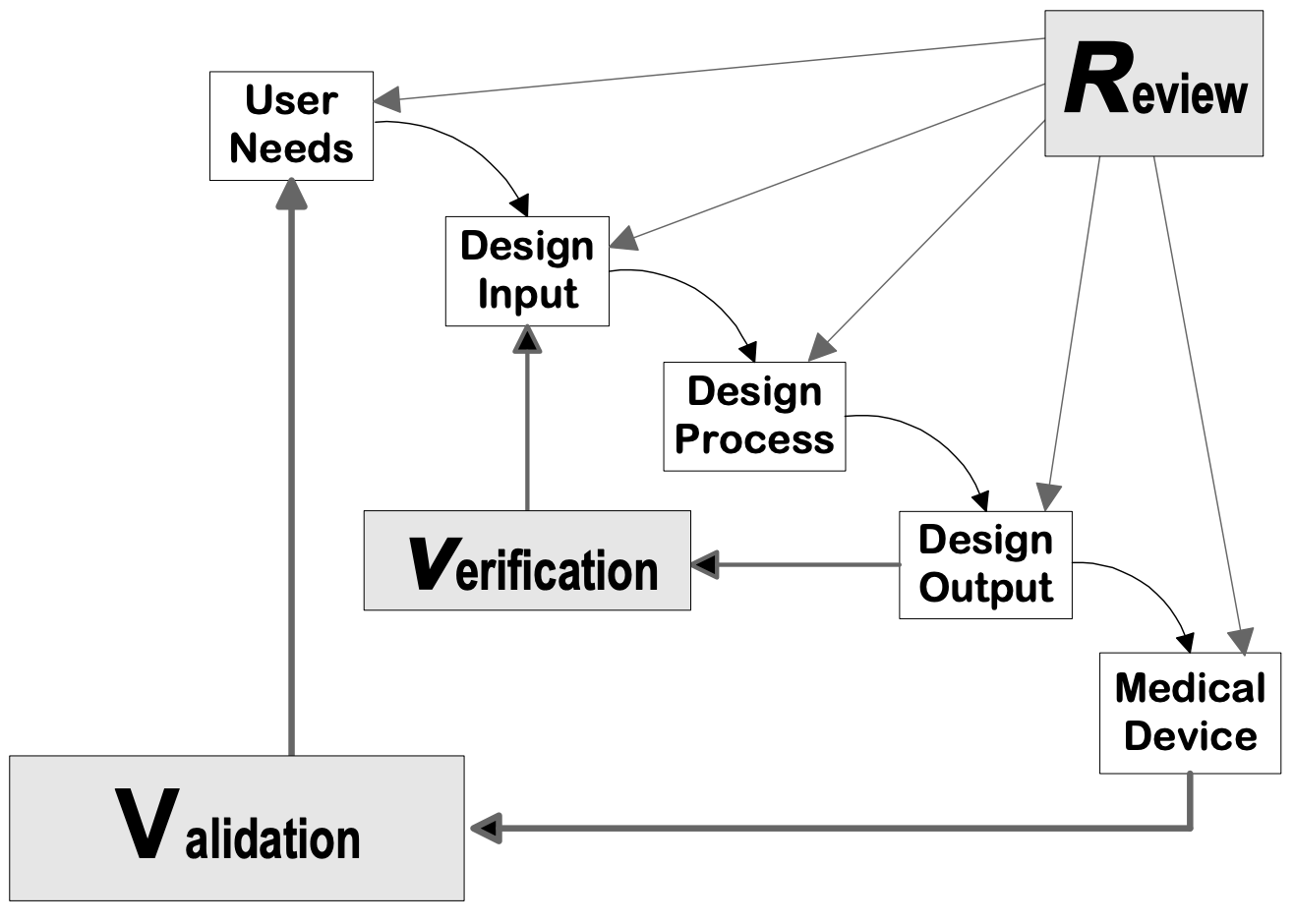
Quelle: Design Control Guidance for Medical Device Manufacturers, CDRH-FDA
Phasen der Geräteentwicklung
D&D Planungsphase
Es sollten D&D-Pläne erstellt und gepflegt werden. Der Plan muss die Entwurfs- und Entwicklungstätigkeiten beschreiben oder darauf verweisen und Verantwortlichkeiten für die Durchführung zuweisen. Die FDA empfiehlt, zumindest die folgenden Punkte in den Plan aufzunehmen:
- Ziele und Aufgaben des D&D-Programms
- Abgrenzung der Verantwortlichkeiten für die Entwurfstätigkeiten
- Ermittlung der wichtigsten Aufgaben und Ergebnisse und Zuweisung der Verantwortung für jede Aufgabe
- Planung der wichtigsten Aufgaben in Übereinstimmung mit dem Zeitplan für die Entwicklung
- Identifizierung der wichtigsten Überprüfungen und Entscheidungspunkte
- Bestimmung der Gutachter, des Gutachterteams und der von den Gutachtern zu befolgenden Verfahren
- Kontrolle der Entwurfsdokumentation
- Benachrichtigungsaktivitäten
Der Plan sollte überprüft, aktualisiert und genehmigt werden, wenn sich Design und Entwicklung weiterentwickeln.
Entwurf Eingabephase
This is the starting point for product design. The medical device is designed and developed to meet the user requirements. Gather the user requirements from various sources such as customer surveys, feedback from the physicians, complaints. These requirements are transferred into design inputs.
- Design-Eingaben: Dies sind die physischen und leistungsbezogenen Anforderungen an ein Gerät, die für den Entwurf des Geräts verwendet werden. Die Phase der Entwurfseingabe besteht aus der Umwandlung von Benutzeranforderungen in Produktanforderungen. Bei der Festlegung der Design-Inputs werden die gesetzlichen Anforderungen berücksichtigt. Die Design-Input-Anforderungen müssen umfassend, eindeutig und objektiv überprüfbar sein. Die Input-Anforderungen können in 3 Kategorien eingeteilt werden:
- Funktionsanforderungen, die beschreiben, was das Gerät tut. Zum Beispiel: Der Rollstuhl muss sich vorwärts bewegen, wenn der Benutzer dies anzeigt.
- Leistungsanforderungen, die angeben, wie viel und wie gut das Gerät funktionieren soll. Zum Beispiel: Der Rollstuhl muss sich mit einer Geschwindigkeit von 2m/s in Vorwärtsrichtung bewegen.
- Schnittstellenanforderungen, spezifizieren Merkmale des Geräts, die für die Kompatibilität mit externen Systemen entscheidend sind, wie z. B. die Benutzer-/Patientenschnittstelle. Zum Beispiel: Der Rollstuhl muss Tasten mit richtungsanzeigenden Symbolen haben
- Hier ist ein Beispiel, das die Bedürfnisse der Benutzer definiert und in Design-Input umwandelt:
- Benutzerbedarf
Das Gerät sollte tragbar und bluetoothfähig sein
- Benutzerbedarf
-
- Entwurfseingabe
Geben Sie die anwendbare, von der FDA anerkannte oder eine internationale Norm an, die eingehalten werden muss. Zum Beispiel:
-
-
- IEEE ANSI C63.27-2017 Amerikanischer nationaler Standard für die Bewertung der drahtlosen Koexistenz
- AAMI TIR 69: Association for the Advancement of Medical Instrumentation – Risikomanagement of Radio-frequency Wireless Coexistence for Medical Devices and Systems (2017)
- IEC 60601-1-2 Ausgabe 3: 2007: Medizinische elektrische Geräte - Teil 1-2: Allgemeine Festlegungen für die Sicherheit - Ergänzungsnorm: Elektromagnetische Verträglichkeit - Anforderungen und Prüfungen
- UL 2054 - Norm für Haushalts- und Gewerbebatterien
-
Führen Sie die Leistung und andere spezifische Eingaben auf. Zum Beispiel:
-
-
- Das Gerät muss mit Gleichstrom betrieben werden.
- Es soll ein Bluetooth-Modul verwendet werden.
- Gewicht: ca. 6lbs oder 6lbs+/- 2lbs (Die Eingaben sollten mit quantitativen Grenzen versehen sein, um eine Überprüfung zu gewährleisten.)
-
Entwurf Ausgabephase
Entwurfsergebnisse sind die Ergebnisse eines Entwurfs in jeder Entwurfsphase und am Ende der gesamten Entwurfsarbeit. Beispiele für Entwurfsergebnisse sind technische Zeichnungen, Beschriftungen, Arbeitsanweisungen und andere Produktspezifikationen. Weitere Entwurfsergebnisse sind Ergebnisse von Risikoanalysen, Ergebnisse von Verifizierungsaktivitäten, Ergebnisse von Biokompatibilitätstests und Software-Quellcode.
Die Konstruktionsergebnisse sollten nicht vor der Überprüfung und Genehmigung durch das zuständige Personal freigegeben werden. Es sollte auch beachtet werden, dass alle Änderungen am Gerät nach der Genehmigung der Auslegungsergebnisse/Eingaben durch die Überprüfung und Genehmigung durch das zuständige Personal kontrolliert werden. Am Ende dieser Phase ist eine Entwurfsprüfung erforderlich.
Phase der Entwurfsprüfung
Nach der Entwurfsphase sollte eine Entwurfsprüfung durchgeführt werden. Die Entwurfsprüfung sollte nach festgelegten Verfahren erfolgen und in der Entwurfsdokumentation (Design History File, DHF) dokumentiert werden. An jeder Entwurfsprüfung sollten Vertreter aus allen Funktionsgruppen teilnehmen.
Es wird empfohlen, formale Überprüfungen am Ende wichtiger Projektmeilensteine durchzuführen. In der Regel werden Design-Reviews nach der Design-Output-Phase, der V&V-Phase und der Design-Transfer-Phase durchgeführt. Dies hängt auch von der Komplexität der Produktentwicklung ab. Die FDA verlangt mindestens eine Entwurfsprüfung.
Entwurfsverifizierungsphase
Die Entwurfsprüfung ist die Bestätigung eines objektiven Nachweises, dass die Entwurfsergebnisse den Entwurfseingaben entsprechen. Im Grunde genommen gilt: Design Input = Design Output. Die Verifizierungsaktivitäten sollten gemäß den festgelegten Verfahren durchgeführt werden. Beispiele für Verifizierungen sind EMV- und elektrische Prüfungen, Sichtprüfungen, nicht-klinische Prüfungen, Fehlerbaumanalysen von Prozessen oder Konstruktionen sowie Fehlerarten- und -auswirkungsanalysen. Die Verifizierung stellt sicher, dass die technischen Anforderungen der Produktspezifikation erfüllt werden. Alle Verifizierungsaktivitäten müssen dokumentiert werden.
Rückverfolgbarkeitsmatrix: Dieses Dokument besteht aus tabellarisch aufgelisteten Entwurfseingaben und -ausgaben. Für jede Eingabe wird auf die entsprechende Ausgabe verwiesen. Diese Überprüfungsmethode wird verwendet, wenn Inputs und Outputs beide Dokumente sind.
Phase der Entwurfsvalidierung
Entwurfsvalidierung bedeutet, dass durch objektive Beweise nachgewiesen wird, dass die Spezifikationen (spezifizierte Anforderungen) mit den Bedürfnissen der Benutzer und dem/den beabsichtigten Verwendungszweck(en) übereinstimmen.
- Prozessvalidierung bedeutet, dass durch einen objektiven Nachweis festgestellt wird, dass ein Prozess durchgängig ein Ergebnis oder Produkt hervorbringt, das den vorgegebenen Spezifikationen entspricht.
- Unter Auslegungsvalidierung versteht man den objektiven Nachweis, dass die Produktspezifikationen mit den Bedürfnissen der Anwender und der beabsichtigten Verwendung(en) übereinstimmen.
Die Validierung ist typischerweise unter realen oder simulierten Bedingungen durchgeführt werden. Examples of validation include klinische Studien, clinical evaluation, human factors tests, address packaging and labeling, analysis and inspections. Results of validation activities and/or validation reports should be documented which will be part of the Design History File (DHF).
Entwurfsübertragungsphase
Nach Abschluss der V&V-Phase erfolgt der Designtransfer. Dazu gehört die Übertragung des Geräteentwurfs in Produktspezifikationen, die die Qualität des Geräts sicherstellen. Diese Phase ist sehr kritisch, denn sobald die Produktion des Geräts beginnt, unterliegt es der Kontrolle von Konstruktionsänderungen und kann bei Problemen zu finanziellen Verlusten führen. Der Designtransfer sollte nach einem festgelegten Verfahren erfolgen. Außerdem sollte sichergestellt werden, dass die Dokumente, die die Produktspezifikationen enthalten, geprüft und genehmigt werden, bevor mit dem Designtransfer begonnen wird.
Phase der Entwurfsänderungen
Die Kontrolle von Konstruktionsänderungen beginnt mit der Übergabe des Entwurfs und setzt sich während des gesamten Lebenszyklus fort. Jede Konstruktionsänderung nach der Konstruktionsübertragung führt zu einer Änderungsmitteilung (Engineering Change Notice, ECN), die nach einem festgelegten Verfahren durchzuführen ist. Es sollte sichergestellt werden, dass bei jeder Konstruktionsänderung auch die zugehörigen Dokumente wie Risikomanagementberichte, Gebrauchsanweisungen, Verifizierungs- und Validierungsberichte überprüft und aktualisiert werden müssen.
Design History File (DHF)
Die DHF ist spezifisch für die US-FDA. Die ISO 13486:2016 verlangt vom Hersteller nicht, eine DHF zu führen.
Für jedes Projekt wird ein Design History File geführt, das alle Ergebnisse jeder Phase enthält. Sie enthält die neuesten Produktinformationen. Die Konstruktions- und Entwicklungsunterlagen müssen leicht zugänglich sein und bei Bedarf abgerufen werden können. In den Konstruktions- und Entwicklungsverträgen sollte das Recht des Herstellers auf Konstruktionsinformationen ausdrücklich festgelegt werden, und es sollten Standards für Form und Inhalt der Konstruktionsunterlagen festgelegt werden.
In der Praxis bieten Designkontrollen Managern und Designern einen besseren Einblick in den Designprozess. Dank der verbesserten Transparenz sind Manager in der Lage, den Designprozess effektiver zu steuern, d. h. Probleme früher zu erkennen, Korrekturen vorzunehmen und die Ressourcenzuweisung anzupassen. Die Konstrukteure profitieren sowohl von einem besseren Verständnis des Konformitätsgrads eines Entwurfs mit den Bedürfnissen der Benutzer und Patienten als auch von einer verbesserten Kommunikation und Koordination zwischen allen am Prozess Beteiligten.
Benötigen Sie Hilfe beim Verständnis und bei der Umsetzung der FDA-Designkontrollen für Ihr Medizinprodukt? Kontakt erfahren Berater für medizinische Geräte auf Kolabtree.
Referenzen:
- FDA, Leitfaden zur Designkontrolle für Hersteller von Medizinprodukten
- Medizinische Regulierungspraktiken, eine internationale Perspektive, Val Theisz
- Designkontrolle, Vortrag von Joseph Tartal
3a. Bundesregister / Bd. 61, Nr. 195
Kolabtree helps businesses worldwide hire freelance scientists and industry experts on demand. Our freelancers have helped companies publish research papers, develop products, analyze data, and more. It only takes a minute to tell us what you need done and get quotes from experts for free.

Unlock Corporate Benefits
• Secure Payment Assistance
• Onboarding Support
• Dedicated Account Manager
Sign up with your professional email to avail special advances offered against purchase orders, seamless multi-channel payments, and extended support for agreements.


