



Shreya Chenni, rédacteur indépendant en matière de réglementation for medical devices, provides a 10-minute guide to FDA design controls for your dispositif médical.
L'une des principales causes des rappels de dispositifs médicaux est le manque de contrôle de la conception, tel qu'identifié par la FDA [3,3a]. Les contrôles de pré-production ont ensuite été ajoutés à la réglementation sur les BPF des dispositifs. Les contrôles de conception sont un ensemble de pratiques et de procédures interdépendantes qui sont intégrées au processus de conception et de développement, c'est-à-dire un système de vérifications et d'équilibres. Les contrôles de conception augmentent la probabilité que la conception transférée à la production se traduise par un dispositif adapté à l'utilisation prévue.
Applicabilité
Tous les dispositifs de classe II, III et les dispositifs de classe I suivants sont soumis à des contrôles de conception :
- Devices automated with computer software
- 868.6810 Cathéter d'aspiration trachéobronchique
- 878.4460 Gant de chirurgien
- 880.6760 Contention protectrice
- 892.5650 Système, applicateur, radionucléide, manuel
- 892.5740 Source, téléthérapie par radionucléides
- Dispositifs automatisés avec des logiciels informatiques
- Cathéters d'aspiration trachéobronchique
- Gants de chirurgien
- Contraintes de protection
- Système, radionucléide, applicateur, manuel
- Source, téléthérapie par radionucléides
Design controls apply to all D&D activities – for novel or improved devices being developed in the pre-market phase, as well as for changes to existing, marketed devices. Design controls do not apply to recherche activities conducted during the proof of concept stage.
Design controls can be applied to any développement de produits process. The following flowcharts are examples of the design controls applied to a traditional Waterfall design process and V-model process for Software (SW).
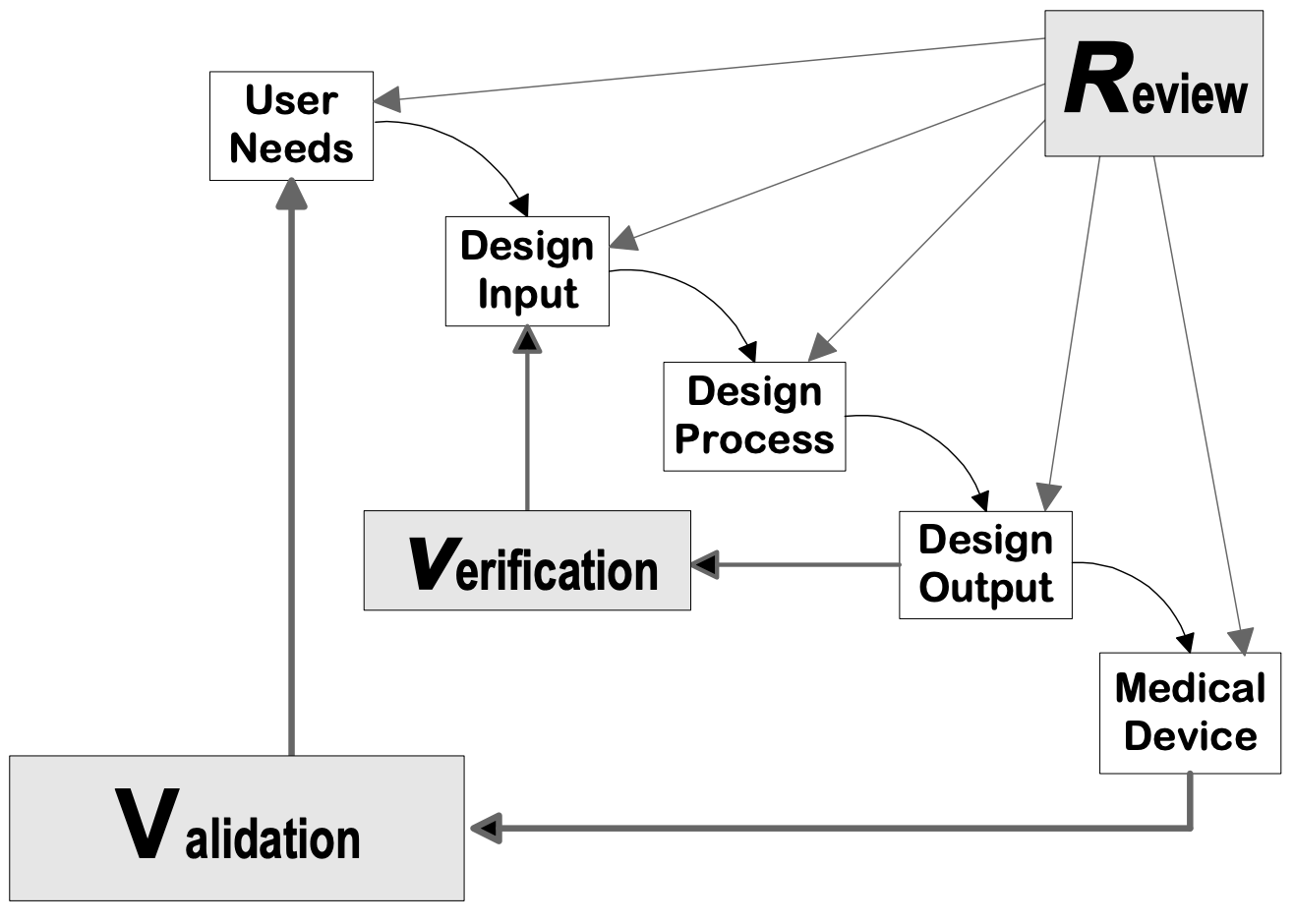
Source : Guide de contrôle de la conception pour les fabricants de dispositifs médicaux, CDRH-FDA
Phases de conception des dispositifs
Phase de planification de la D&D
Des plans de D&D doivent être établis et maintenus. Le plan doit décrire ou faire référence aux activités de conception et de développement et attribuer les responsabilités pour la mise en œuvre. Au minimum, la FDA recommande d'inclure les éléments suivants dans le plan :
- Buts et objectifs du programme de D&D
- Délimitation des responsabilités des activités de conception
- Identifier les principales tâches, les produits livrables et attribuer la responsabilité de chaque tâche.
- Planification des tâches principales en fonction du calendrier de développement principal
- Identifier les principaux examens et points de décision
- Identifier les réviseurs, l'équipe de révision et les procédures à suivre par les réviseurs.
- Contrôles de la documentation de conception
- Activités de notification
Le plan doit être revu, mis à jour et approuvé au fur et à mesure de l'évolution de la conception et du développement.
Phase de saisie des données de conception
This is the starting point for product design. The medical device is designed and developed to meet the user requirements. Gather the user requirements from various sources such as customer surveys, feedback from the physicians, complaints. These requirements are transferred into design inputs.
- Intrants de conception : Il s'agit des exigences physiques et de performance d'un appareil qui sont utilisées pour sa conception. La phase de conception consiste à convertir les exigences de l'utilisateur en exigences du produit. Les exigences réglementaires sont prises en compte lors de la définition des intrants de conception. Les exigences relatives aux intrants de la conception doivent être complètes, non ambiguës et vérifiables objectivement. Les exigences relatives aux intrants peuvent être regroupées en 3 catégories :
- Les exigences fonctionnelles, qui décrivent ce que fait le dispositif. Par exemple : Le fauteuil roulant doit avancer lorsque l'utilisateur l'indique.
- Les exigences en matière de performance, qui spécifieront la quantité et la qualité des performances de l'appareil. Par exemple : Le fauteuil roulant doit se déplacer à une vitesse de 2m/s dans le sens de la marche.
- Les exigences d'interface, spécifient les caractéristiques du dispositif qui sont essentielles à la compatibilité avec les systèmes externes, comme l'interface utilisateur/patient. Par exemple : Le fauteuil roulant doit être équipé de boutons avec des symboles d'indication de direction.
- Voici un exemple, qui définit les besoins des utilisateurs et les convertit en données de conception :
- Besoin de l'utilisateur
Le dispositif doit être portable et compatible Bluetooth
- Besoin de l'utilisateur
-
- Contribution à la conception
Identifiez la norme applicable reconnue par la FDA ou une norme internationale à laquelle vous devez vous conformer. Par exemple :
-
-
- IEEE ANSI C63.27-2017 Norme nationale américaine pour l'évaluation de la coexistence sans fil.
- AAMI TIR 69: Association for the Advancement of Medical Instrumentation – Gestion des risques of Radio-frequency Wireless Coexistence for Medical Devices and Systems (2017)
- IEC 60601-1-2 Edition 3 : 2007 : Appareils électromédicaux - Partie 1-2 : Exigences générales de sécurité - Norme collatérale : Compatibilité électromagnétique - Exigences et essais
- UL 2054 - Norme pour les batteries domestiques et commerciales
-
Dressez la liste des performances et autres données spécifiques. Par exemple :
-
-
- Le dispositif doit être alimenté en courant continu
- Le module Bluetooth doit être utilisé
- Poids : environ 6lbs ou 6lbs+/- 2lbs (Les entrées doivent avoir des limites quantitatives pour assurer la vérification)
-
Phase de sortie de la conception
Les produits de la conception sont les résultats d'un effort de conception à chaque phase de la conception et à la fin de l'effort total de conception. Les dessins techniques, l'étiquetage, les instructions de travail et autres spécifications de produits sont des exemples de produits de conception. D'autres produits de conception comprennent les résultats de l'analyse des risques, les résultats des activités de vérification, les résultats des tests de biocompatibilité et le code source des logiciels.
Les sorties de conception ne doivent pas être publiées avant d'être examinées et approuvées par le personnel responsable. Il convient également de noter que toute modification apportée au dispositif après l'approbation des sorties/entrées de conception sera contrôlée par la révision et l'approbation du personnel concerné. Une revue de la conception est nécessaire à la fin de cette phase.
Phase de révision de la conception
Une revue de conception doit être organisée après la phase de production de la conception. Les revues de conception doivent suivre les procédures établies et être documentées dans le dossier historique de la conception (DHF). Les participants à chaque revue de conception doivent être des représentants de tous les groupes fonctionnels.
Il est recommandé d'effectuer des revues formelles à la fin des étapes importantes du projet. En général, les revues de conception sont effectuées après la phase de sortie de la conception, la phase de V&V et la phase de transfert de la conception. Cela dépend également de la complexité du développement du dispositif. La FDA exige au moins une revue de conception.
Phase de vérification de la conception
La vérification de la conception est la confirmation par des preuves objectives que les résultats de la conception correspondent aux données d'entrée de la conception. En gros, c'est Design Input = Design Output. Les activités de vérification doivent être réalisées conformément aux procédures établies. Les exemples de vérification comprennent les essais CEM et électriques, l'inspection visuelle, les activités d'essais non cliniques, l'analyse par arbre de défaillance du processus ou de la conception et l'analyse des modes de défaillance et de leurs effets. La vérification permet de s'assurer que les exigences techniques de la spécification du produit sont respectées. Toutes les activités de vérification doivent être documentées.
Matrice de traçabilité : Ce document se compose d'entrées et de sorties de conception répertoriées sous forme de tableaux. Pour chaque entrée, la sortie correspondante est référencée. Cette méthode de vérification est utilisée lorsque les entrées et les sorties sont toutes deux des documents.
Phase de validation de la conception
La validation de la conception consiste à établir par des preuves objectives que les spécifications (exigences spécifiées) sont conformes aux besoins des utilisateurs et à l'utilisation prévue.
- La validation d'un processus consiste à établir par des preuves objectives qu'un processus produit de façon constante un résultat ou un produit conforme à ses spécifications prédéterminées.
- Validation de la conception : établir par des preuves objectives que les spécifications du dispositif sont conformes aux besoins de l'utilisateur et à l'utilisation prévue.
La validation est généralement conduite dans des conditions réelles ou simulées. Examples of validation include les essais cliniquesL'évaluation clinique, les tests de facteurs humains, l'emballage et l'étiquetage des adresses, l'analyse et les inspections. Les résultats des activités de validation et/ou les rapports de validation doivent être documentés et faire partie du dossier historique de la conception (DHF).
Phase de transfert de la conception
Après l'achèvement de l'étape V&V, le transfert de la conception a lieu. Il s'agit de transférer la conception du dispositif dans les spécifications du produit afin de garantir la qualité du dispositif. Cette phase est très critique car une fois que la production du dispositif commence, il sera soumis au contrôle des modifications de conception et peut entraîner des pertes financières si des problèmes sont rencontrés. Le transfert de la conception doit se faire selon la procédure établie. Il faut également s'assurer que les documents qui comprennent les spécifications du produit sont examinés et approuvés avant de lancer le transfert de la conception.
Phase de modifications de la conception
Le contrôle des modifications de la conception commence avec le transfert de la conception et se poursuit tout au long du cycle de vie. Toute modification de la conception après le transfert de la conception donne lieu à un avis de modification technique (ECN) qui doit être exécuté selon une procédure établie. Il convient de s'assurer qu'avec tout changement de conception, les documents connexes tels que le rapport de gestion des risques, les instructions d'utilisation, les rapports de vérification et de validation doivent être réexaminés et mis à jour également.
Dossier historique de conception (DHF)
Le DHF est spécifique à la FDA américaine. La norme ISO 13486:2016 n'impose pas au fabricant de tenir à jour un DHF.
Un dossier sur l'historique de la conception est tenu à jour pour chaque projet et comprend tous les produits livrables de chaque phase. Il comprend les dernières informations sur les produits. Les documents de conception et de développement doivent être facilement disponibles et accessibles en cas de besoin. Les contrats de conception et de développement doivent spécifier explicitement le droit du fabricant aux informations de conception et établir des normes pour la forme et le contenu des documents de conception.
Dans la pratique, les contrôles de conception offrent aux gestionnaires et aux concepteurs une meilleure visibilité du processus de conception. Grâce à cette visibilité accrue, les gestionnaires sont en mesure de diriger plus efficacement le processus de conception, c'est-à-dire de reconnaître les problèmes plus tôt, d'apporter des corrections et d'ajuster l'affectation des ressources. Les concepteurs bénéficient à la fois d'une meilleure compréhension du degré de conformité d'une conception aux besoins des utilisateurs et des patients, et d'une amélioration de la communication et de la coordination entre tous les participants au processus.
Vous avez besoin d'aide pour comprendre et mettre en œuvre les contrôles de conception de la FDA pour votre dispositif médical ? Contactez une personne expérimentée consultants en dispositifs médicaux sur Kolabtree.
Références :
- FDA, Design Control guidance for Medical Device Manufacturers (guide de contrôle de la conception pour les fabricants de dispositifs médicaux)
- Pratiques de réglementation médicale, une perspective internationale, Val Theisz
- Contrôle de la conception, présentation par Joseph Tartal
3a. Federal Register / Vol. 61, No. 195
Kolabtree helps businesses worldwide hire freelance scientists and industry experts on demand. Our freelancers have helped companies publish research papers, develop products, analyze data, and more. It only takes a minute to tell us what you need done and get quotes from experts for free.

Unlock Corporate Benefits
• Secure Payment Assistance
• Onboarding Support
• Dedicated Account Manager
Sign up with your professional email to avail special advances offered against purchase orders, seamless multi-channel payments, and extended support for agreements.


