



Sundeep Agarwal, medical device regulatory consultant on Kolabtree, shares the essential requirements of a FDA 510k premarket notification to ensure success.
With the current $156 billion market size, and expected $208 billion [1] by 2023, the US medical device market is undoubtedly lucrative. Additionally, the aging population, history of chronic diseases, the healthcare system and disruption in supply chain encourages global manufacturer to invest and expand their business horizon in the United States. Like any other global manufacturer, if you think you can leave an imprint in the world largest medical device market, you have just hit on the right blog to prepare and understand the regulatory requirement with respect to FDA’s 510(k)for your device which is utmost vital for the application process.
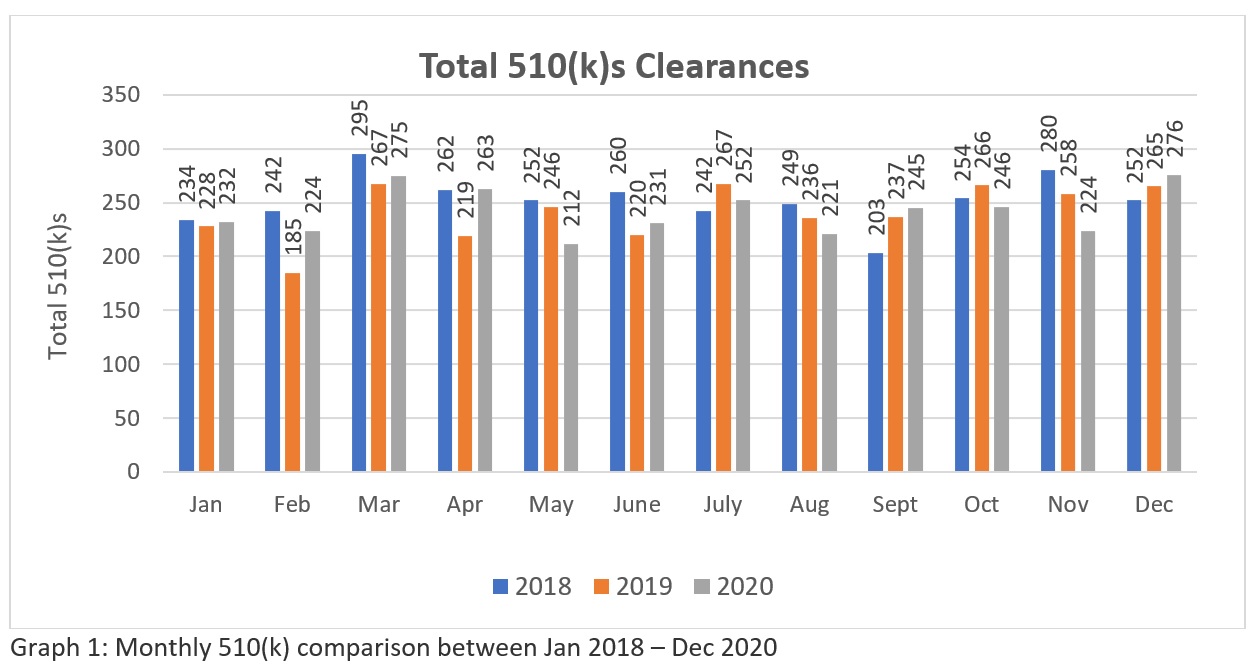
A trend analysis of three years FDA’s 510k clearances data (Source: https://www.fda.gov/medical-devices/device-approvals-denials-and-clearances/510k-clearances) reveal that a total of 3025-510(k)swere cleared between January 2018 to December 2018, 2894 – 510(k)s between January 2019 to December 2019 and 2901 – 510(k)s cleared between January 2020 to December 2020. These figures (Refer Table 1) suggest that currently, FDA has the potential to clear more than two hundred 510(k) per month for the industry to meet the healthcare market needs in the US. While the graphical representation (Refer Graph 1)indicates about FDA’s consistency in their review approach amidst the global pandemic. Although the number is not significantly big in comparison to large number of applications made globally. Hope FDA will increase its resources in the future . Therefore, manufacturers require a careful preparation and submission to assure they get through in their first attempt.
| Year 2018 | Year 2019 | Year 2020 | |
| Total 510(k)s | 3025 | 2894 | 2901 |
| Total with summaries | 2885 | 2735 | 2759 |
| Total with statements | 140 | 159 | 142 |
Table 1: The table indicates yearly 510(k) cleared between Jan 2018 – Dec 2020
FDA 510k requirements: Finding the keys
Unless well versed with the FDA’s 510(k) preparation and submission process, it can really be stressful and challenging. If a device manufacturer intends to market a device in the US that is either Class I, II, and III but not an exempt from a 510(k) or which does not require a Premarket Approval application (PMA) then its eligible for a 510(K). Proceeding further, manufacturer should have a clarity on the terms ‘predicate device’ and ‘substantial equivalence (SE)’. A legally marketed devices in the US to which a manufacturer wants to claim an equivalence is generally known as a predicate device. On the other hand, Substantial equivalence means establishing an evidence-based comparison for a new device that a manufacturer wants to place in the US market is safe and effective as the existing predicate device is particularly with respect to same intended use, technological characteristics. In case if the new device differs in technological characteristics, it shouldn’t differ in terms of safety and efficacy and the manufacturer is able to demonstrate that the device is as safe and effective as the existing legally marketed device.
Unlocking the process
Some of the core factors for a successful 510(k) involves correct product code classification, identification and availability of a predicate device in the US market, well-planned substantial equivalence comparison with evidences, a robust quality management system, design controls, strict adherence to the current FDA forms and best utilization of refuse to accept (RTA) checklist (also refer figure 1). The Refuse to accept (RTA) checklist guides on the various acceptance criteria that FDA follows while performing a substantive review of a 510(k). For all three types of 510(k), there are different type of RTA available. RTA Checklist can be accessed from here.
Manufacturer should note that it’s not the number of pages or merely submitting the test results or studies would get them a 510(k) clearance rather a well explained, sequential write-up (as recommended by FDA in their guidance document) based on scientific justification and evidences that will be carefully reviewed by the FDA within a fixed timeframe ultimately achieves the 510(k). Majority of 510(k) are rejected because of lack of clarity or inadequate evidence to claim a substantial equivalence [as in Traditional 510(k)]or insufficient comparison to FDA recognised conformity standards [as in Abbreviated 510(k)]. It is observed that failure to manage design controls and lack of a well-established quality system is another big reason for such a failure. Lastly unavailability of a competent team or lack of clear understanding about the regulation is undeniable factor that contributes to the rejection in addition to the above.

Figure 1: Key points to consider for a Premarket Notification 510(k)
Types of FDA 510(k) Premarket Notification
In general, there are three types of 510(k)s that a manufacturer may submit to the FDA (Refer figure 2). They are:
(1) Traditional 510(k) – Most common and majority of the 510(k) are in this application type,
(2) Special 510(k) – Required only when a changes are made inlabel(s) or design or certain changes in indication for use in an existing previously cleared device. The content should meet the requirements as defined in 21 CFR Part 807.87 and 21 Part 807.90,
(3) Abbreviated 510(k) – Applies in case a manufacturer is able to produce reports on the use of special control or guidance’s or declaration of conformity based on FDA’s recognised standards.

Figure 2: Types of Premarket Notification 510(k)
The 510k submission process
Prior to a submission, manufacturer will have to register their organisation with the FDA. The process is term as establishment registration as per 21 CFR Part 807 after a payment of fees directly to the FDA and the same shallbe renewed annually. For the current financial year 2021, the fee is $ 5,546 for an establishment registration. Once should check the exact fees by visiting the FDA user fee programs official website.
FDA recommends 20 sections in a Traditional or Abbreviated 510(k) but not necessarily all sections shall be applicable to a manufacturer. At times, if an information in a particular section does not apply to their device, they may include the section heading and write “This section does not apply” or “N/A” under the same. The core recommended section of the 510(k) as advised in the guidance document by FDA are listed below:
- Medical Device User Fee Cover Sheet (Form FDA 3601): Indicates a receipt of a user fee paid to the FDA by the manufacturer.
- Center for Devices and Radiological Health (CDRH) Premarket Review Submission Cover Sheet (Form FDA 3514): This is a voluntary form to provide all sort of administration information to the FDA about the organisation and submission.
- A 510(k) Cover Letter: A description about the purpose, content and administrative information about the 510(k) should be incorporated in this letter. It is recommended to refer Appendix A of “Format for Traditional and Abbreviated 510(k)s Guidance for Industry and Food and Drug Administration Staff; dtd September 13, 2019”.
- Indications for Use Statement (Form FDA 3881): It should be uniform in the entire 510(k). It should also define if the device is to be marketed as prescription or over-the-counter (OTC) use.
- 510(k) Summary or 510(k) Statement: To be prepared in accordance with 21 CFR part 807. Here the manufacturer is expected to summarise the 510(k) and incorporate information about the rest of the content.
- Truthful and Accuracy Statement: It is a statement by an authorised person of the organisation certifying that all information submitted to the FDA related to the 510(k) is truthful and accurate.
- Class III Summary and Certification: Applicable to only Class III devices. It’s a summary of safety and effectiveness and an assurance that a reasonable search has been carried out and the manufacturer has all relevant safety information based on similar marketed devices.
- Financial Certification or Disclosure Statement: If a manufacturer submits clinical evidence, a disclosure statement by the clinical investigator is to be enclosed. FDA form 3454 or Form 3455 may be refered.
- Declarations of Conformity and Summary Reports: Here information related to use of voluntary consensus standards or the basis of general use of such standards shall be provided.
- Device Description: A brief description of the device design, models or accessories are to be included section.
- Executive Summary/Predicate Comparison: A short description of the device, indications for use and technology along with device comparison table is recommended in this section.
- Substantial Equivalence Discussion: A detailed comparison between manufacturer device with the predicate device to demonstrate the substantial equivalence.
- Proposed Labelling: It will include proposed labelling for the medical device as per 21 CFR 807.87(e) or as per 21 CFR 809.10 requirements in case of an in vitro diagnostic device.
- Sterilization and Shelf Life: The method of sterilization, relevant validation, and claimed shelf life to be included in this section.
- Biocompatibility: Studies protocol, reports and assurance that the biocompatibilities studies were performed following Good Laboratory Practices. FDA recommends use of ISO 10993 for biocompatibility studies.
- Software: If the device incorporates software, this section shall be applicable.
- Electromagnetic Compatibility and Electrical Safety: Applicable mainly for electrical or active devices. FDA recommends use of ANSI/AAMI (ES) 60601-1for general safety testing or an equivalent method.
- Performance Testing – Bench: It is recommended to include the various performance test conducted by the manufacturer or in a third-party lab which may include but not limited to mechanical or engineering or biological test results.
- Performance Testing – Animal: If animal studies were conducted and is included in the submission, FDA recommends to describe the tests and provide the results that support the performance characteristics.
- Performance Testing – Clinical: If the submission included clinical data/studies, FDA expects the inclusion of information about the clinical study protocol and objective, test methods, study endpoints and statistical tools used in the clinical study.
Please be aware that there is no standard template or all-in-one ready to fill in 510(k) application format; though FDA’s 21 CFR Part 807, Subpart-E describes the procedure and guides a manufacturer about the establishment registration and device listing. And further several relevant forms associated with such a submission which are helpful to prepare can be downloaded from the given FDA’s official link i.e. https://www.fda.gov/medical-devices/premarket-notification-510k/510k-forms . It is advisable to refer the various FDA’s guidance’s on 510(k), available in public domain in order to meet the regulatory compliances and document accordingly.Although it’s not a practice to perform 510(k) pre-clearance facility inspections, but manufacturer shall implement a robust quality system as per 21 CFR Part 820 requirement and be inspection ready, just in case it takes place.
There is also a provision known as “Third Party Review Program” for certain low to moderate risk devices. As the name suggest, it’s not the FDA that directly reviews the 510(k) application, but accredited third party organisation approved by the FDA does the job. Based on the review and recommendation made by the accredited third party, FDA concludes a decision about a 510(k) clearance. Currently there are 10 such third-party organisation available. For a submitter or manufacturer, it will wise to check if their device is eligible for a third-party review at https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfpcd/classification.cfm
510k Application Timeline
A submitter or manufacturer while submitting a 510(k) to FDA also requires to prepare an eCopy of their 510(k) and submit the same to CDRH or CBER at document control center (DCC). Based on the submitted information, manufacturer will be asked for additional information during the different review stage (Refer figure 3).
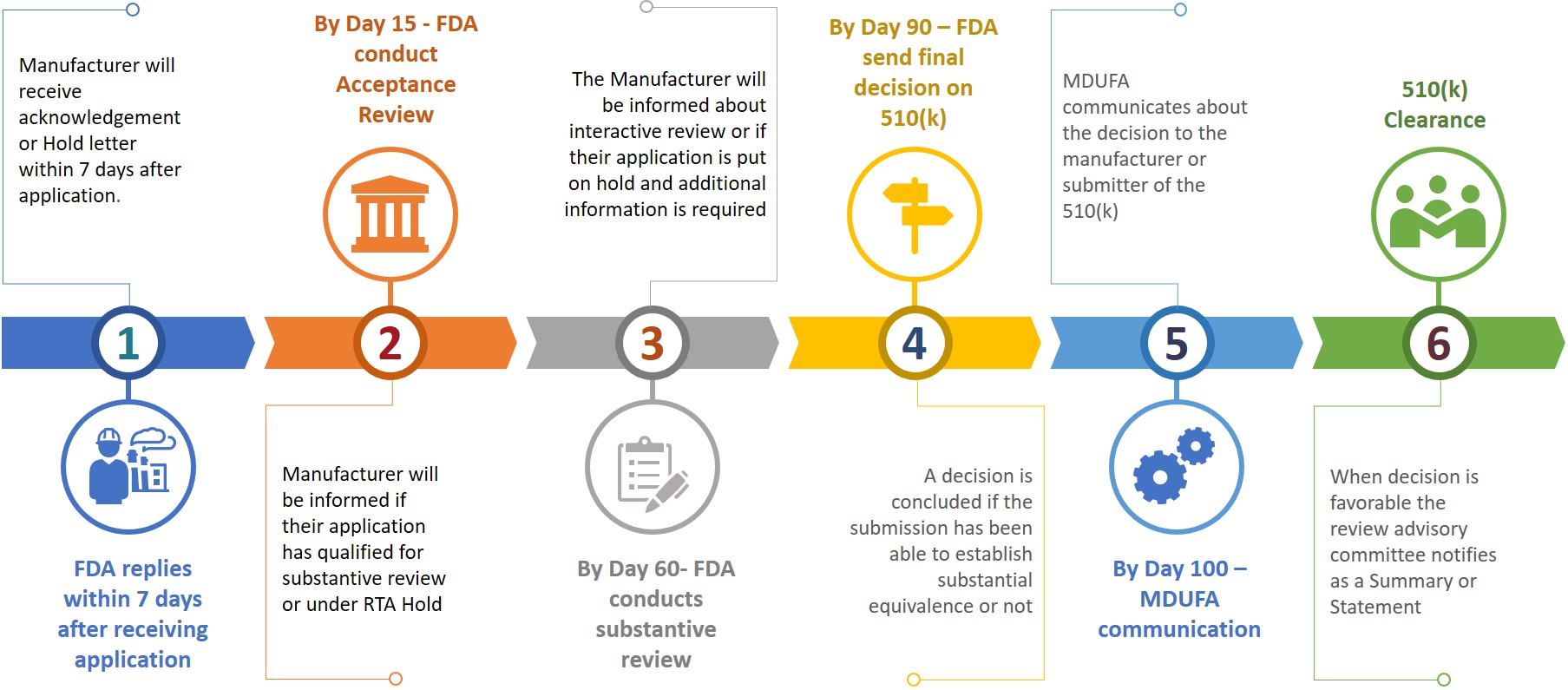
Figure 3: Tentative review timeline
The submitter will have 180 calendar days to respond when they receive an RTA hold or if FDA wants additional information. Not resolving any issues within allowed 18 calendar days will result in auto deletion from the review system or shall be considered withdrawn. In an event of deletion or withdrawn of a 510(k), the submitter will have to re-apply a new application after paying the necessary fee while the K number may be quoted in the new application for the same device. A 510(k) clearance can be achieved within 100 days after submission while it may even take 6 – 9 months to get the clearance.
References
- The 510(k) Program: Evaluating Substantial Equivalence in Premarket Notifications [510(k)] Guidance for Industry and Food and Drug Administration Staff Document issued on: July 28, 2014.
- Format for Traditional and Abbreviated 510(k)s Guidance for Industry and Food and Drug Administration Staff; Document issued on September 13, 2019.
- [1]The medical technology spotlight (US) overview, prepared in collaboration with the International Trade Administration’s Industry & Analysis Unit (I&A) https://www.selectusa.gov/medical-technology-industry-united-states (Last accessed on June 11, 2021)

About the author
Sundeep Agarwal, Subject Matter Expert and FDA, CE (MDR & IVDR) Consultant
With a decade of experience, he is globally sought-after Leader, Speaker & Consultant in the field of QA & RA, Quality Management System, Product Design & Development, Risk Management, Commercial Scale-up, Industrial Manufacturing and Clinical Studies of medical devices.An active member of a Technical Group (Software as Medical Device) at Asian Harmonization Working Party.He joins Medical Device industry/government, collaborated conferences a speaker and panelist frequently on ISO 13485, EU MDR, IVDR, CE Certification, CER, PMS, USFDA, 510(K), ISO 14971, MDSAP, Combination Devices, Artificial Intelligence , etc. He prominently serves as a guest lecturer in various MBA and Pharmacy educational institutions in India. Contact him directly for a project on Kolabtree.
Kolabtree helps businesses worldwide hire freelance scientists and industry experts on demand. Our freelancers have helped companies publish research papers, develop products, analyze data, and more. It only takes a minute to tell us what you need done and get quotes from experts for free.

Unlock Corporate Benefits
• Secure Payment Assistance
• Onboarding Support
• Dedicated Account Manager
Sign up with your professional email to avail special advances offered against purchase orders, seamless multi-channel payments, and extended support for agreements.


