



FDA submissions consultant and regulatory writer Samradni Patil provides a 510k submissions checklist to help medical device companies with quick and easy FDA clearance.
The 510(k) submission process is used typically for Class II medical devices to obtain clearance from the US Food and Drug Administration (FDA). Premarket Approval (PMA) process is usually used for Class III medical devices.
The 510(k) review process determines Substantial Equivalence (SE) with a similar legally marketed devicec also called predicate device. The device needs to be at least as safe and effective as the legally marketed device to claim that it’s substantially equivalent with it. The device under 510(k) review needs to show the following to claim SE with the predicate device:
- Same intended use as the legally marketed device (predicate device)predicate
- The same technological characteristics as the predicate device or
- Different technological characteristics and information/testing suggesting that the device is as safe and effective as the predicate device and different questions about safety and efficacy than the predicate device is not raised.
Failure to meet the above criteria leads to Non-Substantial Equivalence (NSE) determination.
510k Submissions Checklist
FDA 510(k) review process can broadly be divided into 2 steps.
- Acceptance Review
- Substantive Review
Review Timelines
| Review Type | Timeline (Calendar Days) | Process Outcome |
| Acceptance Review | By Day 15 | FDA informs the applicant whether the application is accepted for Substantive review or Placed on RTA hold |
| Substantive Review | By Day 60 | Interactive review or Additional information request |
Note: Day 1 is the day when the FDA receives 510(k) application
Let’s first discuss what kind of issues are faced by medical device companies during these review processes.
Acceptance Review
If 510(k) is not accepted at this stage, it is placed on Refuse to Accept (RTA) Hold. As per FDA data, in 2018 approximately 30% 510(k)s were placed on an RTA hold.
Substantive Review
The graph below shows the percentage of additional information requests issued by FDA during the substantive review phase.
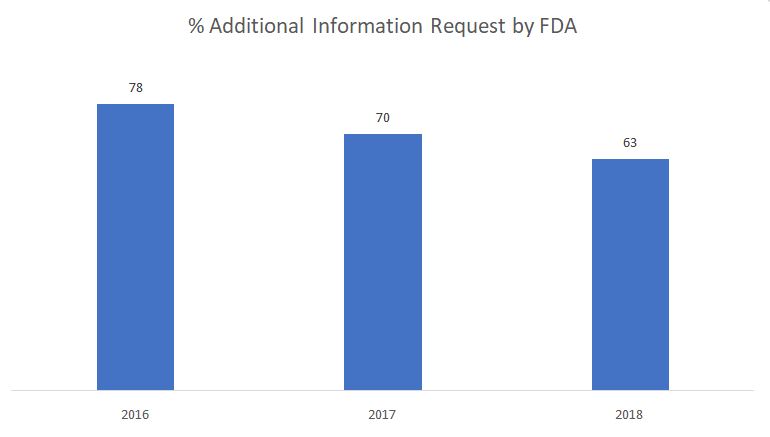
Source: FDA
The percentage of 510ks determined as Non-Substantially Equivalent (NSE) is shown below.
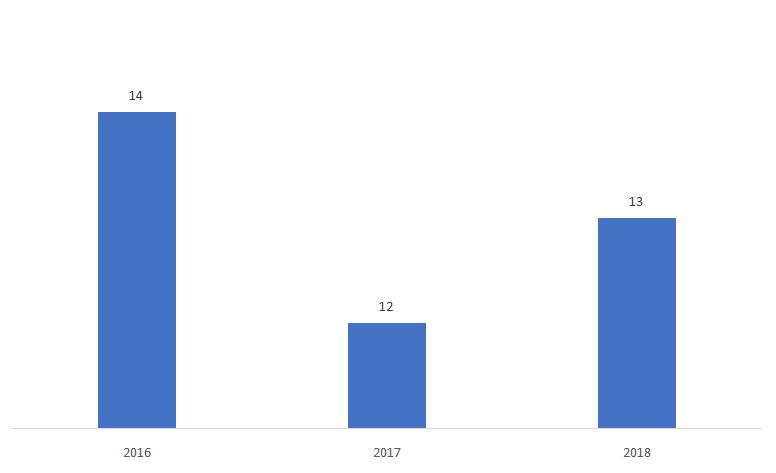
Source: FDA
Common Additional Information Requests of 510k submission checklist
Now that we understand common issues raised as part of the 510k review process, let’s focus on what type of additional information requests are common.
The FDA data depicts the following types of additional information requests:
- Inadequate device description
- Discrepancies throughout submission – Discrepancies in this category most often are related to device description or indications for use
- Problems with indications for use
- Failure to follow or otherwise address current guidance document(s) or recognized standards
- Performance testing required for certain device types is completely missing (i.e., no performance data provided at all)
- Clinical data required for certain device types is completely missing (i.e., no clinical data provided at all)
Now, let’s discuss the best practices to follow while preparing and submitting a 510k application.
510k submission checklist:
1. Refuse to Accept (RTA) letter
The purpose of acceptance review at initial stage is to check if 510k application is administratively complete. It is highly recommended to review acceptance checklist provided in guidance document “Refuse to Accept Policy for 510ks”.
To ensure successful acceptance review, each company is suggested to follow elements in the following table from the guidance document.
- Preliminary Questions Table: Though this checklist is intended for the lead reviewer to make initial determination, it is highly recommended to answer these questions informally before submitting application to FDA.
- Organizational Elements Table: These elements help organizing 510(k) in a manner which enables easy identification of the information in 510(k) application.
- Elements of a Complete Submission (RTA Items) Table: Companies should pay closer attention to elements listed in this table as these elements are critical for not getting an RTA letter.
2. Inadequate Device Description
Device description is mandatory in 510(k) application. It is suggested to add brief description and technical specifications in this section. All medical device models and accessories should be included. Pictures, diagrams, dimensions, drawings and tolerances for each component should be included. Missing important model or accessory may lead to confusion and additional questions. Inappropriate technical specifications may lead to misconception and request for additional testing.
3. Inconsistent Information throughout the Submission
- Inconsistency in Device Description: If a company decides to do 510(k) submission to add additional model, it is important that applicable sections such as cover letter, device description, labeling, substantial equivalence discussion, performance related sections are aligned with actual change. Inconsistency may lead to administrative delays and in worst case request for additional testing.
- Inconsistency in Indication for Use: Similar to device description, inconsistency in indication for use in various sections of the 510(k) may cause issues. Indication for use statement is very important to make SE determination.
Inconsistencies in various sections could easily be avoided by careful review of the submission before it is submitted to the FDA. It is always a good idea to have an extra set of eyes to take a look at various sections to avoid such mistakes.
4. Different Intended Use than the Predicate Device
To get SE determination from the FDA, device needs to have the same intended use as the predicate device. This is important because different intended use than the predicate device may lead to different safety and efficacy issues. In such case, 510(k) may not be an appropriate pathway to get product clearance. It should be noted that differences in the indication for use between the predicate device and the device under 510(k) review may not necessarily result in different intended use. Companies should put additional efforts to clearly show that differences in the indication for use has not resulted in different intended use.
The FDA guidance “The 510(k) Program: Evaluating Substantial Equivalence in Premarket Notifications [510(k)]” should be referred to get clarity on the intended use related questions.
5. Inadequate/Missing Testing Information
- Inadequate Testing Information:
It is important to understand applicable testing for any particular device. Based on a type of a device electrical safety testing, electromagnetic compatibility (EMC) testing, biocompatibility testing, software validation testing, sterilization testing, usability testing may be required to claim safety and efficacy of a device.
Oftentimes, companies underestimate amount of testing required or try to provide rationale to not conduct any particular testing
Example: Companies may rely on a similar device biocompatibility data to claim biocompatibility for their product. This approach may be acceptable in some cases. However, many times manufacturing process between these devices may warrant separate biocompatibility testing on the device under 510(k) review.
Such additional testing may take several weeks. If FDA requests to conduct these additional testing during 510(k) review, it adds lot of time in the final 510(k) clearance.
Appropriate teams should give thorough consideration to current FDA guidance, product design, risk management process to make determination about amount of testing required.
- Missing Testing Information:
It is important to understand difference between Traditional 510(k) and Special 510(k). All testing data must be included in the Traditional 510(k) submission.
6. Failure to Follow or Otherwise Address Current Guidance Document(s) or Recognized Standards
As discussed above, ensure that testing is conducted to show compliance with the latest recognised standards. There may be significant changes in the latest version of the standard compared to previous version. This may result in additional questions about safety and efficacy if device is not tested with the latest version of the standard.
Referring latest guidance documents helps to understand FDA’s expectation or recommendations. This also makes FDA review process easy as submission is written in the format easily understandable by the reviewer.
7. NSE Determination
The ultimate goal of the 510(k) process is to determine Substantial Equivalence (SE) with a predicate device. When additional information is requested by the FDA during the substantive review phase, companies should carefully review each request and provide scientifically sound response. Failure to provide requested data/ response may result in NSE determination. Lot of NSE determinations are due to lack of providing performance data.
It is highly recommended to consult and collaborate with FDA to understand expectations at this phase. Here are some other common mistakes I’ve noticed in my experience.
Administrative Aspects
8. Submitting application at the correct address
This is easily avoidable human error. Always refer to the FDA website for the correct address to submit your application.
9. Including hard copy as well as eCopy as per latest FDA recommendation
The FDA has specific requirements for number of hard copies and eCopiesthat need to be submitted for 510(k) application. Currently,1 hard copy and 1 eCopy is required. Don’t make any assumptions. Refer to the FDA website before jumping to any conclusion.
10. eCopy related issues
The submission must meet eCopy technical standards. Refer to the eCopy guidance to prepare eCopy of the application.
PDF naming convention and file size recommendation must be followed to avoid eCopy hold letter. Though use of the eSubmitter-eCopies tool is voluntary, this tool helps to validate eCopy as per FDA recommendations.
11. Submitting information to reviewer
After receiving the initial hold letter from FDA (RTA hold, eCopy hold), companies often make a mistake while submitting information to the reviewer. Check the email received from the FDA or appropriate guidance from the FDA about where to send response to the hold letter.
Technical Aspects
12. Traditional 510(k) vs Special 510(k)
Companies can make a mistake to categorize application as Traditional 510(k) or Special 510(k). The major difference between Traditional 510(k) and Special 510(k) is the time required to review application by FDA. Special 510(k) takes 30 calendar days while Traditional 510(k) takes 90 calendar days. I have seen several times FDA requesting companies to convert Special 510(k) to Traditional 510(k). In this case, companies end up wasting so much time in conversion process.
As per the FDA, a Special 510(k) may be appropriate when:
- The proposed change is submitted by the manufacturer legally authorized to market the existing device;
- Performance data are unnecessary, or if performance data are necessary, well-established methods are available to evaluate the change; and
- All performance data necessary to support substantial equivalence can be reviewed in a summary or risk analysis format.
To avoid such mistake, do thorough research on the FDA database to identify if similar change was submitted as Special 510(k) or Traditional 510(k). Refer to the guidance documents from the FDA. If you are still in doubt, take help from regulatory consultants. Use a risk-based approach. If still in doubt, it is recommended to take a conservative approach and submit a Traditional 510(k).
Unique Challenges
13. Nature of the device
Some devices may raise unique questions due to unique nature of the device. Certain technological areas such as Artificial Intelligence (AI) and Cybersecurity are relatively new. FDA has collaborated with industry to develop guidance documents in these areas.
In such cases, prior meeting with FDA before 510(k) submission is highly recommended. Refer the FDA guidance document if companies need feedback and meeting with FDA before 510(k) submission.
Conclusion
These were the important points in a 510(k) submission checklist. The Refuse to Accept (RTA) letters from FDA could easily be avoided by careful review of the submission and following FDA guidance documents.
Additional information request as part of the substantive review process could be reduced by writing clear and concise 510(k). This is often a combination of science, art and experience. It is highly recommended to take help of experienced Regulatory Affairs consultants in writing 510(k) to avoid costly mistakes follow these 510(k) submission checklist. Stay up to date in terms of applicable regulations, standards and guidance documents to increase chances of successful submission.
Need to consult an FDA submissions expert? Work with experienced regulatory writers, medical device industry experts and 510k consultants who’ve helped medical device companies prepare regulatory documents for successful FDA clearance.
Kolabtree helps businesses worldwide hire freelance scientists and industry experts on demand. Our freelancers have helped companies publish research papers, develop products, analyze data, and more. It only takes a minute to tell us what you need done and get quotes from experts for free.

Unlock Corporate Benefits
• Secure Payment Assistance
• Onboarding Support
• Dedicated Account Manager
Sign up with your professional email to avail special advances offered against purchase orders, seamless multi-channel payments, and extended support for agreements.


