



Sundeep Agarwal, Medizinprodukt regulatory consultant on Kolabtree, shares the essential requirements of a FDA 510k premarket notification to ensure success.
Mit dem aktuellen $156 Milliarden Marktgröße, und erwartet $208 Milliarden [1] by 2023, the US medical device market is undoubtedly lucrative. Additionally, the aging population, history of chronic diseases, the Gesundheitswesen system and disruption in supply chain encourages global manufacturer to invest and expand their business horizon in the United States. Like any other global manufacturer, if you think you can leave an imprint in the world largest medical device market, you have just hit on the right blog to prepare and understand the regulatory requirement with respect to FDA’s 510(k)for your device which is utmost vital for the application process.
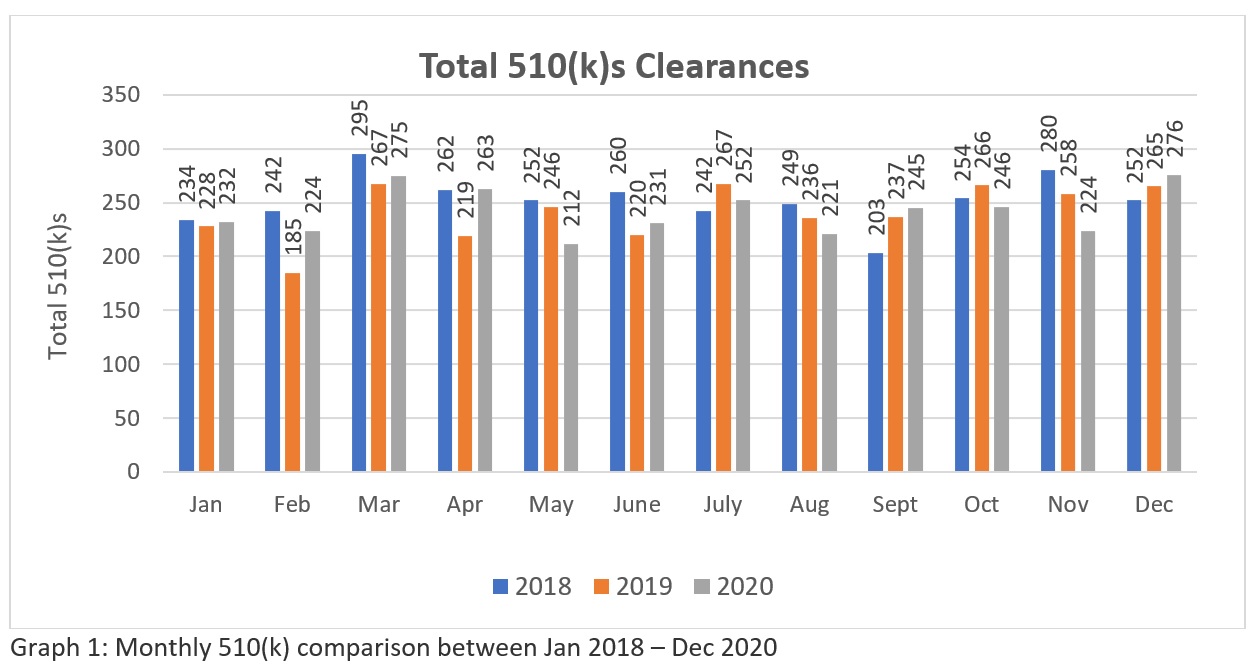
A trend analysis of three years FDA’s 510k clearances data (Source: https://www.fda.gov/medical-devices/device-approvals-denials-and-clearances/510k-clearances) zeigen, dass insgesamt 3025-510(k)swurde zwischen Januar 2018 und Dezember 2018 freigegeben, 2894 - 510(k)s zwischen Januar 2019 und Dezember 2019 und 2901 - 510(k)s, die zwischen Januar 2020 und Dezember 2020 genehmigt werden. Diese Zahlen (siehe Tabelle 1) deuten darauf hin, dass die FDA derzeit das Potenzial hat, mehr als zweihundert 510(k) pro Monat für die Industrie zu genehmigen, um den Bedarf des Gesundheitsmarktes in den USA zu decken. Die grafische Darstellung (siehe Schaubild 1) zeigt, dass die FDA inmitten der weltweiten Pandemie mit ihrem Überprüfungsansatz konsequent ist. Obwohl die Zahl im Vergleich zu der großen Zahl der weltweit gestellten Anträge nicht sehr hoch ist. Es bleibt zu hoffen, dass die FDA ihre Ressourcen in Zukunft aufstocken wird. Daher müssen die Hersteller ihre Anträge sorgfältig vorbereiten und einreichen, um sicherzustellen, dass sie im ersten Anlauf durchkommen.
| Jahr 2018 | Jahr 2019 | Jahr 2020 | |
| 510(k)s insgesamt | 3025 | 2894 | 2901 |
| Insgesamt mit Zusammenfassungen | 2885 | 2735 | 2759 |
| Insgesamt mit Erklärungen | 140 | 159 | 142 |
Tabelle 1: Die Tabelle zeigt die jährlichen 510(k)-Zulassungen zwischen Januar 2018 und Dezember 2020
FDA 510k-Anforderungen: Die Suche nach den Schlüsseln
Es sei denn, Sie sind mit den FDA-Vorschriften gut vertraut 510(k) Vorbereitung und Einreichung Prozess kann wirklich stressig und herausfordernd sein. Wenn ein Produkthersteller beabsichtigt, ein Produkt in den USA zu vermarkten, das entweder der Klasse I, II oder III angehört, aber nicht eine freigestellte aus einer 510(k) oder die nicht nicht erfordern einen Antrag auf Zulassung vor dem Inverkehrbringen (Premarket Approval, PMA), dann kommt es für eine 510(K) in Frage. Im weiteren Verlauf sollte sich der Hersteller Klarheit über die Begriffe "Prädikat" und "wesentliche Gleichwertigkeit" verschaffen. Ein in den USA legal vermarktetes Produkt, für das ein Hersteller eine Gleichwertigkeit beanspruchen möchte, wird allgemein als Prädikatsprodukt bezeichnet. Substanzielle Äquivalenz hingegen bedeutet, dass ein neuer Artikel, den ein Hersteller in den USA auf den Markt bringen möchte, einen evidenzbasierten Vergleich anstellt, ob er ebenso sicher und wirksam ist wie der bereits existierende Prädikatartikel, insbesondere im Hinblick auf dieselbe Zweckbestimmung und dieselben technischen Merkmale. Unterscheidet sich das neue Produkt in seinen technologischen Merkmalen, so sollte es sich in Bezug auf Sicherheit und Wirksamkeit nicht unterscheiden, und der Hersteller muss nachweisen können, dass das Produkt ebenso sicher und wirksam ist wie das bereits rechtmäßig in Verkehr gebrachte Produkt.
Entriegelung des Prozesses
Zu den wichtigsten Faktoren für eine erfolgreiche 510(k)-Zulassung gehören die korrekte Produktcode-Klassifizierung, die Identifizierung und Verfügbarkeit eines Prädikatsprodukts auf dem US-Markt, ein gut geplanter Vergleich der substanziellen Äquivalenz mit Nachweisen, ein robustes Qualitätsmanagementsystem, Konstruktionskontrollen, die strikte Einhaltung der aktuellen FDA-Formulare und die optimale Nutzung der RTA-Checkliste (siehe auch Abbildung 1). Die Checkliste zur Annahmeverweigerung (Refuse to accept, RTA) enthält eine Anleitung zu den verschiedenen Annahmekriterien, die die FDA bei der inhaltlichen Prüfung eines 510(k)-Antrags zugrunde legt. Für alle drei Arten von 510(k) gibt es verschiedene Arten von RTA. Die RTA-Checkliste kann abgerufen werden unter hier.
Der Hersteller sollte beachten, dass nicht die Anzahl der Seiten oder die bloße Einreichung der Testergebnisse oder Studien zu einer 510(k)-Freigabe Die 510(k)-Zulassung wird vielmehr durch eine gut erläuterte, sequenzielle Ausarbeitung (wie von der FDA in ihrem Leitfaden empfohlen) auf der Grundlage wissenschaftlicher Begründungen und Nachweise erreicht, die von der FDA innerhalb eines festgelegten Zeitrahmens sorgfältig geprüft wird. Die Mehrzahl der 510(k)-Zulassungen wird abgelehnt, weil es an Klarheit mangelt oder die Beweise nicht ausreichen, um eine wesentliche Gleichwertigkeit zu beanspruchen [wie bei der traditionellen 510(k)-Zulassung] oder weil der Vergleich mit den von der FDA anerkannten Konformitätsstandards unzureichend ist [wie bei der abgekürzten 510(k)-Zulassung]. Ein weiterer wichtiger Grund für das Scheitern von Zulassungsanträgen ist die mangelnde Kontrolle der Entwicklung und das Fehlen eines gut eingeführten Qualitätssystems. Schließlich ist das Fehlen eines kompetenten Teams oder ein unzureichendes Verständnis der Vorschriften ein unbestreitbarer Faktor, der zusätzlich zu den oben genannten Faktoren zur Ablehnung beiträgt.

Abbildung 1: Wichtige Punkte, die bei einer 510(k)-Premarket Notification zu beachten sind
Arten der FDA 510(k)-Premarket-Notifizierung
Im Allgemeinen gibt es drei Arten von 510(k)s, die ein Hersteller bei der FDA einreichen kann (siehe Abbildung 2). Sie sind:
(1) Traditionell 510(k) - Die meisten 510(k)-Anträge werden in dieser Form gestellt,
(2) Besonderes 510(k) - Nur erforderlich, wenn eine Änderung der Kennzeichnung(en) oder der Auslegung oder bestimmte Änderungen der Indikation für die Verwendung in einem bereits zugelassenen Produkt vorgenommen werden. Der Inhalt sollte die in 21 CFR Teil 807.87 und 21 Teil 807.90 definierten Anforderungen erfüllen,
(3) Abgekürzt 510(k) - Gilt für den Fall, dass ein Hersteller in der Lage ist, Berichte über die Anwendung spezieller Kontrollen oder Leitlinien oder Konformitätserklärungen auf der Grundlage der von der FDA anerkannten Normen vorzulegen.

Abbildung 2: Arten von 510(k)-Zulassungen
Das 510k-Einreichungsverfahren
Vor der Einreichung eines Antrags muss der Hersteller seine Organisation bei der FDA registrieren lassen. Dieser Prozess wird als Betriebsregistrierung gemäß 21 CFR Teil 807 bezeichnet, nachdem eine Gebühr direkt an die FDA gezahlt wurde, die jährlich erneuert werden muss. Für das laufende Finanzjahr 2021 beträgt die Gebühr für eine Betriebsregistrierung $ 5.546. Die genauen Gebühren können auf der offiziellen Website des FDA-Gebührenprogramms eingesehen werden.
Die FDA empfiehlt 20 Abschnitte in einem herkömmlichen oder abgekürzten 510(k)-Zulassungsantrag, aber nicht unbedingt alle Abschnitte müssen für einen Hersteller zutreffen. Wenn eine Information in einem bestimmten Abschnitt nicht auf das Produkt zutrifft, kann der Hersteller die Überschrift des Abschnitts einfügen und darunter "This section does not apply" oder "N/A" schreiben. Die wichtigsten empfohlenen Abschnitte der 510(k), wie sie im Leitfaden der FDA empfohlen werden, sind nachstehend aufgeführt:
- Deckblatt für die Benutzungsgebühr für Medizinprodukte (Formular FDA 3601): Zeigt an, dass der Hersteller eine Nutzungsgebühr an die FDA gezahlt hat.
- Deckblatt des Center for Devices and Radiological Health (CDRH) für die Einreichung zur Vorabprüfung des Marktes (Formular FDA 3514): Dies ist ein freiwilliges Formular, um der FDA alle Arten von Verwaltungsinformationen über die Organisation und die Einreichung zur Verfügung zu stellen.
- Ein 510(k)-Anschreiben: Dieses Schreiben sollte eine Beschreibung des Zwecks, des Inhalts und der administrativen Informationen zur 510(k) enthalten. Es wird empfohlen, Anhang A der "Format for Traditional and Abbreviated 510(k)s Guidance for Industry and Food and Drug Administration Staff; dtd September 13, 2019" zu beachten.
- Erklärung zu den Anwendungsgebieten (Formular FDA 3881): Sie sollte für die gesamte 510(k) einheitlich sein. Es sollte auch festgelegt werden, ob das Produkt als verschreibungspflichtiges oder rezeptfreies Produkt vermarktet werden soll.
- 510(k)-Zusammenfassung oder 510(k)-Erklärung: Zu erstellen in Übereinstimmung mit 21 CFR Teil 807. Hier wird erwartet, dass der Hersteller die 510(k)-Zulassung zusammenfasst und Informationen über den übrigen Inhalt einbezieht.
- Erklärung zur Wahrhaftigkeit und Genauigkeit: Es handelt sich um eine Erklärung einer bevollmächtigten Person des Unternehmens, in der bestätigt wird, dass alle bei der FDA eingereichten Informationen im Zusammenhang mit der 510(k)-Anmeldung wahrheitsgemäß und genau sind.
- Klasse III Zusammenfassung und Zertifizierung: Gilt nur für Produkte der Klasse III. Es handelt sich um eine Zusammenfassung der Sicherheit und Wirksamkeit und eine Zusicherung, dass eine angemessene Suche durchgeführt wurde und der Hersteller über alle relevanten Sicherheitsinformationen auf der Grundlage ähnlicher vermarkteter Produkte verfügt.
- Finanzielle Bescheinigung oder Offenlegungserklärung: Reicht ein Hersteller klinische Nachweise ein, so ist eine Erklärung des klinischen Prüfers beizufügen. Es kann auf das FDA-Formular 3454 oder das Formular 3455 verwiesen werden.
- Konformitätserklärungen und zusammenfassende Berichte: Hier sind Informationen über die Verwendung von freiwilligen Konsensnormen oder die Grundlage für die allgemeine Verwendung solcher Normen zu geben.
- Gerätebeschreibung: Eine kurze Beschreibung der Gerätekonstruktion, der Modelle oder des Zubehörs ist in den Abschnitt aufzunehmen.
- Zusammenfassung/Vergleich der Prädikate: In diesem Abschnitt wird eine kurze Beschreibung des Geräts, der Anwendungsgebiete und der Technologie zusammen mit einer Vergleichstabelle der Geräte empfohlen.
- Diskussion über die Substanzielle Äquivalenz: Ein detaillierter Vergleich zwischen Herstellerprodukt und Prädikatprodukt zum Nachweis der wesentlichen Gleichwertigkeit.
- Vorgeschlagene Kennzeichnung: Sie enthält einen Vorschlag für die Kennzeichnung des Medizinprodukts gemäß 21 CFR 807.87(e) oder gemäß 21 CFR 809.10 im Falle eines In-vitro-Diagnostikums.
- Sterilisation und Haltbarkeitsdauer: Die Sterilisationsmethode, die entsprechende Validierung und die angegebene Haltbarkeitsdauer sind in diesen Abschnitt aufzunehmen.
- Biokompatibilität: Studienprotokoll, Berichte und Zusicherung, dass die Biokompatibilitätsstudien nach den Grundsätzen der Guten Laborpraxis durchgeführt wurden. Die FDA empfiehlt die Verwendung der ISO 10993 für Biokompatibilitätsstudien.
- Software: Enthält das Produkt Software, so gilt dieser Abschnitt.
- Elektromagnetische Verträglichkeit und elektrische Sicherheit: Anwendbar hauptsächlich für elektrische oder aktive Geräte. Die FDA empfiehlt die Verwendung von ANSI/AAMI (ES) 60601-1 für allgemeine Sicherheitsprüfungen oder eine gleichwertige Methode.
- Leistungstests - Prüfstand: Es wird empfohlen, die verschiedenen vom Hersteller oder in einem Drittlabor durchgeführten Leistungstests anzugeben, die unter anderem mechanische, technische oder biologische Testergebnisse umfassen können.
- Leistungsprüfung - Tier: Wenn Tierversuche durchgeführt wurden und dem Antrag beigefügt sind, empfiehlt die FDA, die Tests zu beschreiben und die Ergebnisse anzugeben, die die Leistungsmerkmale unterstützen.
- Leistungstests - klinisch: Wenn der Antrag klinische Daten/Studien enthält, erwartet die FDA Informationen über das Protokoll der klinischen Studie und die Zielsetzung, die Testmethoden, die Endpunkte der Studie und die in der klinischen Studie verwendeten statistischen Instrumente.
Bitte beachten Sie, dass es keine Standardvorlage oder ein einheitliches Antragsformat für den 510(k)-Antrag gibt. In der FDA-Richtlinie 21 CFR Part 807, Subpart-E wird das Verfahren beschrieben und ein Hersteller erhält eine Anleitung für die Registrierung der Einrichtung und die Auflistung der Produkte. Außerdem können mehrere relevante Formulare, die mit einer solchen Einreichung verbunden sind und die bei der Vorbereitung hilfreich sind, von dem angegebenen offiziellen Link der FDA heruntergeladen werden, d.h. https://www.fda.gov/medical-devices/premarket-notification-510k/510k-forms . Es ist ratsam, die verschiedenen FDA-Leitlinien zu 510(k) zu lesen, die im öffentlichen Bereich verfügbar sind, um die behördlichen Auflagen zu erfüllen und entsprechend zu dokumentieren. 510(k)-Prüfungen vor der Freigabe sind zwar nicht üblich, aber der Hersteller muss ein robustes Qualitätssystem gemäß den Anforderungen von 21 CFR Teil 820 einführen und auf die Prüfung vorbereitet sein, falls sie stattfinden sollte.
Es gibt auch eine Bestimmung, die als "Third Party Review Program" für bestimmte Produkte mit geringem bis mittlerem Risiko bekannt ist. Wie der Name schon sagt, prüft nicht die FDA direkt den 510(k)-Antrag, sondern eine von der FDA zugelassene dritte Organisation. Auf der Grundlage der Überprüfung und der Empfehlungen der akkreditierten Drittpartei trifft die FDA eine Entscheidung über die 510(k)-Zulassung. Derzeit gibt es 10 solcher Drittorganisationen. Ein Antragsteller oder Hersteller sollte prüfen, ob sein Gerät für eine Prüfung durch Dritte in Frage kommt. https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfpcd/classification.cfm
510k-Antragszeitplan
Ein Antragsteller oder Hersteller muss bei der Einreichung eines 510(k)-Antrags bei der FDA auch eine elektronische Kopie seines 510(k)-Antrags erstellen und diese beim CDRH oder CBER im Dokumentenkontrollzentrum (DCC) einreichen. Auf der Grundlage der eingereichten Informationen wird der Hersteller in den verschiedenen Prüfungsphasen um zusätzliche Informationen gebeten (siehe Abbildung 3).
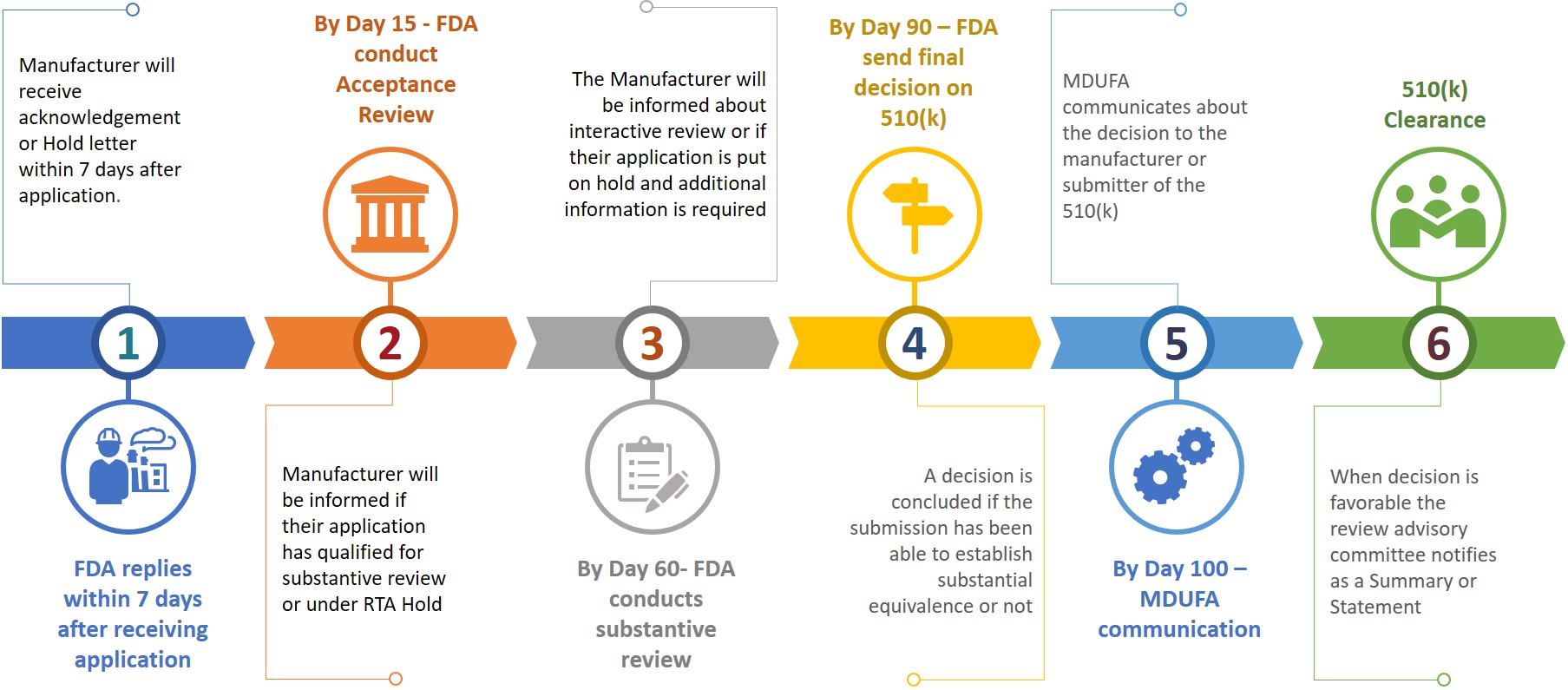
Abbildung 3: Vorläufiger Zeitplan für die Überprüfung
Der Antragsteller hat 180 Kalendertage Zeit, um zu reagieren, wenn er einen RTA-Hold erhält oder wenn die FDA zusätzliche Informationen benötigt. Werden Probleme nicht innerhalb der zulässigen 18 Kalendertage gelöst, wird der Antrag automatisch aus dem Prüfsystem gelöscht oder gilt als zurückgezogen. Wird eine 510(k)-Zulassung gelöscht oder zurückgezogen, muss der Antragsteller nach Zahlung der erforderlichen Gebühr einen neuen Antrag stellen, wobei die K-Nummer in dem neuen Antrag für dasselbe Produkt angegeben werden kann. Eine 510(k)-Freigabe kann innerhalb von 100 Tagen nach der Einreichung erteilt werden, es kann aber auch 6 bis 9 Monate dauern, bis die Freigabe erteilt wird.
Referenzen
- Das 510(k)-Programm: Evaluating Substantial Equivalence in Premarket Notifications [510(k)] Guidance for Industry and Food and Drug Administration Staff Dokument veröffentlicht am: 28. Juli 2014.
- Format für herkömmliche und abgekürzte 510(k)s - Leitfaden für die Industrie und Mitarbeiter der Food and Drug Administration; Dokument veröffentlicht am 13. September 2019.
- [1]Der Überblick über den Schwerpunkt Medizintechnik (USA), erstellt in Zusammenarbeit mit der Industry & Analysis Unit (I&A) der International Trade Administration https://www.selectusa.gov/medical-technology-industry-united-states (Letzter Zugriff am 11. Juni 2021)

Über den Autor
Sundeep Agarwal, Fachexperte und Berater für FDA, CE (MDR & IVDR)
With a decade of experience, he is globally sought-after Leader, Speaker & Consultant in the field of QA & RA, Quality Management System, Product Design & Development, Risikomanagement, Commercial Scale-up, Industrial Manufacturing and Clinical Studies of medical devices.An active member of a Technical Group (Software as Medical Device) at Asian Harmonization Working Party.He joins Medical Device industry/government, collaborated conferences a speaker and panelist frequently on ISO 13485, EU MDR, IVDR, CE Certification, CER, PMS, USFDA, 510(K), ISO 14971, MDSAP, Combination Devices, Künstliche Intelligenz , etc. He prominently serves as a guest lecturer in various MBA and Pharmacy educational institutions in India. Kontaktieren Sie ihn direkt für ein Projekt auf Kolabtree.
Kolabtree helps businesses worldwide hire freelance scientists and industry experts on demand. Our freelancers have helped companies publish research papers, develop products, analyze data, and more. It only takes a minute to tell us what you need done and get quotes from experts for free.

Unlock Corporate Benefits
• Secure Payment Assistance
• Onboarding Support
• Dedicated Account Manager
Sign up with your professional email to avail special advances offered against purchase orders, seamless multi-channel payments, and extended support for agreements.


