



Nare Simonyan, freelance regulatory affairs specialist at Kolabtree, provides a comprehensive guide to a regulatory dossier and its format.
Introduction: What is a regulatory dossier?
Regulatory dossier is a package of documents, which may include all required information regarding newly developed drug products and/or generics, which is required by EU and US regulatory authorities for granting marketing authorization approvals. The main information that is included in the package is administrative information, data related to the quality, safety and efficacy of drug product, which can be submitted by CTD (Common Technical Document) format both paper and electronic version. Since the information submitted in paper format was enormous, agencies are now encouraging applications to be submitted in eCTD format.
CTD (Common Technical Document)
In 2003, the members of ICH (International Council of Harmonization) have agreed to assembly all quality, safety and efficacy information in a common format which used to be called CTD. The CTD is a format/structure for Modules 1 through 5 of the NDA (New Drug Application), MAA (Marketing Authorization Application), and global medicinal applications. Module 1 contains administrative regional information which is differ for each country. Modules 2, 3, 4 and 5 are common for all regions. The CTD structure applies to both investigational and commercial applications (IND (Investigational New Drug) and NDA (US); IMPD (Investigational Medicinal Product Dossier) and MAA (EU), and global applications).
The CTD was primarily used for new marketing applications such as NDA, BLA (Biologics License Application), MAA, NDS (New Drug Substance), JNDA (Japanese New Drug Application), etc. With additional guidance and guidelines from the FDA and EMA, the CTD is now required for all applications, including those for clinical trials—IMPD and INDs. All Drug Master Files (DMF) and Active Substance Master Files (ASMF) must follow the structure of the CTD. The process of submitting regulatory dossier is regulated by Code of Federal Regulations (CFR) (Law in US) and Directives (Law in EU).
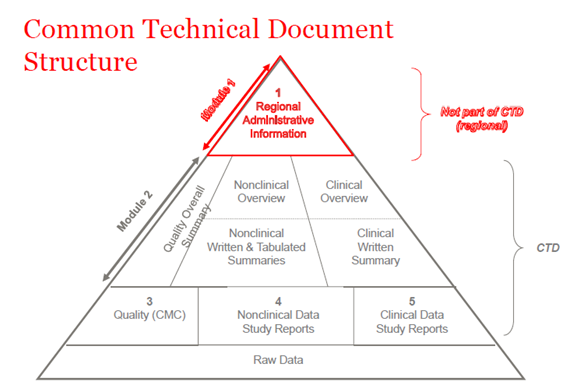
Figure 1. The structure of CTD
CTD-Module 1
Specifications for US Module 1: Administrative Information and Prescribing Information
e-CTD Module 1 section contains administrative and labeling documents. All applications and related submissions have the same organizational structure for documents of Module 1.
Please see below some specifications on documents to be provided within Module 1:
Cover Letter (Section 1.2)
Cover letters contain pertinent information which aid communication within the review process. Following information is recommended to be included in cover letter (please refer to “Electronic Common Technical Document (eCTD) v4.0 TECHNICAL CONFORMANCE GUIDE”)
- Regulatory description of the submission, including appropriate regulatory information, and any desired hyperlinks to submitted information
- Technical description of the submission, including the approximate size of the submission (e.g., 2 gigabytes)
- Statement that the submission is virus free, with a description of the software (name, version, and company) that was used to check the files for viruses
- A regulatory and technical point of contact for the submission, including email address.
Field Copy Certification (Section 1.3)
Field Copy Certification should be included within eCTD for marketing applications. It can be a letter to District office notifying that eCTD submission will be submitted to FDA. The letter should include:
- Drug and application number
- FDA center and division
- Application is in eCTD format.
References (Section 1.4)
References may contain the following sub-sections (additional content can be required, as well)
- Letters of Authorization (LOAs)
- Cross Referencing Previously Submitted Information that is not in eCTD Format
Information Amendments (Section 1.11)
This can be used for submission of responses to Information Requests (IR), where the information being submitted does not fit under any heading in Module 2, 3, 4 or 5. If the IR response impacts documents submitted in Modules 2 – 5, the new or replacement documents should be submitted to the appropriate location in Module 2 – 5 and referenced in the document from Section 1.11.
Marketing Annual Reports (Section 1.13)
For each study or trial described in the post marketing requirement/commitments files a bookmark should be included.
Labeling (Section 1.14)
1.14 section describes how to provide specific labeling documents. It may include Draft labeling, Final labeling, Listed drug labeling, Investigational drug labeling, Foreign labeling and Product labeling for 2253 submissions*. Provided information may contain the history, content and samples of labeling.
Form FDA 2253*: Transmittal of Advertisements and Promotional Labeling for Drugs and Biologics for Human Use
Advertisements and Promotional Labeling Material (Section 1.15)
Advertisements and promotional labeling materials are restricted in US, it should be reflected to requirements mentioned in FDA Guidance Providing Regulatory Submissions in Electronic and Non-Electronic Format—Promotional Labeling and Advertising Materials for Human Prescription Drugs.
For promotional materials submitted as part of the post marketing reporting requirements, hypertext links to references or labeling suggested to be provided. References improve the efficiency of a review.
Risk Evaluation and Mitigation Strategy (REMS) (Section 1.16)
REMS supplement is intended to propose a new REMS or modifications (major and/or minor) to an approved REMS. In appropriate FDA Form submission type REMS supplement need to be selected. REMS assessments, REMS revisions, and REMS correspondences are not supplements. In this case submission type should be selected “other” when filling the FDA Form.
Specifications for European Union (EU) Module 1: Administrative Information and Prescribing Information
Cover Letter (Section 1.0)
For each application a cover letter should be provided. “Notes to Reviewers” documents can be included as Appendix to the cover letter, in case further information need to be provided in order to facilitate navigation.
Comprehensive Table of Contents (Section 1.1)
A comprehensive table of contents should be provided for each type of application, which may contain all module sections that have been submitted as part of concerned application. In case of new applications, all sections should be addressed.
Application Form (Section 1.2)
Depending on the type of the submission relevant application form should be included in regulatory dossier.
The different application forms are available on the Website of the European Commission /
DG Enterprise:
- New Applications and Extension Applications http://ec.europa.eu/enterprise/pharmaceuticals/eudralex/homev2.htm#2b
- Variation applications
http://ec.europa.eu/enterprise/pharmaceuticals/eudralex/homev2.htm#2c
- Renewal applications
http://ec.europa.eu/enterprise/pharmaceuticals/eudralex/homev2.htm#2c
Product Information (Section 1.3)
Section 1.3 contains information regarding
- Summary of Product Characteristics (SPC), labelling and package leaflet (PIL) (Section 1.3.1)
- Mock-up (Section 1.3.2)
- Specimen (Section 1.3.3)
- Consultation with Target Patient Groups (Section 1.3.4)
- Product Information already approved in the Member States (Section 1.3.5) (where applicable)
- Braille (Section 1.3.6)
Information about the Experts (Section 1.4)
In this section it is necessary to include information regarding experts with providing detailed reports of the documents and information which constitute Modules 3, 4, and 5 in accordance with Article 12 of Directive 2001/83 / EC. For additional part of this section can be used a signed expert reports for the different scientific parts of the dossier. The signed Expert Reports requirements presented below:
- The Quality Overall Summary, Non-clinical Overview / Summary and Clinical Overview / Summary in Module 2,
- A declaration signed by the experts in Module 1.4.
- A brief information on the educational background, training and occupational experience in Module 1.4.
The relevant expert declaration(s) must be provided for post-authorization applications.
In cases where marketing authorization holders wish to distinguish such declaration from any
previous declarations, the relevant procedure number of the reference member state/EMEA
may be included on top.
Sub-sections of Information about the Experts in presented below:
- Quality (Section 1.4.1)
- Non-Clinical (1.4.2)
- Clinical (Section1.4.3)
For more information and relevant templates please refer to Article 12 and in accordance with Annex I, Part I 1.4 of Directive 2001/83/EC.
Specific requirements for Different Types of Applications (Section 1.5)
Information for Bibliographical Applications (Section 1.5.1)
Based upon Article 10a of Directive 2001/83/EC applicants should provide a concise document, summarizing the grounds and evidence used for demonstrating that the constituent(s) of the medicinal product have a well-established use, taking into consideration acceptable level of safety and efficacy, as outlined in Part II.1 of Annex I to Directive 2001/83/EC.
Information for Generic, ‘Hybrid’ or Bio-similar Applications (Section 1.5.2)
Based upon Article 10(1), 10(3) or 10(4) of Directive 2001/83/EC, applicants should provide here a concise document, summarizing the grounds and evidence used for demonstrating the type of medicinal product for which an application is submitted. Medicinal products can be generics, hybrid products, and biosimilars.
(Extended) Data / Market Exclusivity (Section 1.5.3)
Section 1.5.3 is required when MA holder/applicant would like to claim (additional) data/market exclusivity while applying for a new indication or change in classification. In this case relevant legal provisions and requirements must be considered.
Exceptional Circumstances (Section 1.5.4)
According to Article 22 of Directive 2001/83/EC and Article 14(7) of Regulation (EC) No 726/2004, an authorization can be granted in case of exceptional circumstances, when specific procedures, in particular concerning the safety of the medicinal product, notification to the competent authorities of any incident relating to its use are introduced by applicant. Only for objective and verifiable exceptions an authorization may be granted.
Conditional Marketing Authorization (Section 1.5.5)
Mentioned section is applicable for centralized procedure. References for this section are Article 14(7) of Regulation (EC) No 726/2004 and “Guideline on the scientific application and the practical arrangements on the Conditional Marketing Authorization”.
Environmental Risk Assessment (Section 1.6)
In accordance with Article 8 (ca) and (g) of Directive 2001/83/EC any potential risk of the medicinal product to the environment must be considered by applicant while applying for marketing authorization approval. The requirements in the Directive are related to use, storage and disposal of medicinal products, and they are not applicable to the synthesis or manufacture of the product. Applications for marketing authorizations for medicinal products which do not contain GMOs (Non-GMO Section 1.6.1) and do contain GMOs (GMO 1.6.2) should include an indication of any potential risks in Module 1 of regulatory dossier.
For more information please refer to “Guideline on the Environmental Risk Assessment for medicinal products for human use”
Information relating to Orphan Market Exclusivity (Section 1.7)
This section is applicable only for orphan medicines. Required information on details and procedure are present in “European Commission guideline on aspects of the application of Article 8 of Regulation (EC) No 141/2000: Assessment of similarity and/or clinical superiority of orphan medicinal products when assessing marketing authorization applications and variations.”
Section 1.7.1 Similarity and Section 1.7.2 Market Exclusivity are applicable, if orphan medicinal product has been authorized for the condition which covers the proposed therapeutic indication being applied for, and a period of market exclusivity is in force, applicants should provide a critical report addressing the possible similarity with the authorized orphan medicinal product and concluding on similarity or “non” similarity (1.7.1). If the medicinal product, which is the subject of the application for marketing authorization is deemed to be “similar” to an orphan medicinal product covered by the above-mentioned market exclusivity provisions, the applicant must furthermore provide justification that one of the derogations laid down as described in Article 8.3, paragraphs (a) to (c) of Regulation (EC) No 141/2000(1.7.2).
Information relating to Pharmacovigilance (Section 1.8)
Pharmacovigilance System (Section 1.8.1)
Pharmacovigilance is the science and activities related to detecting, assessing, understanding, and preventing adverse effects or any other drug-related problems.
This applies throughout the life cycle of medicine equally to the pre-approval stage as to the post-approval. The pharmacovigilance system is a very important section for applying marketing authorization.
Detailed description of pharmacovigilance system must be provided according to Article 8 (ia) of Directive 2001/83/EC. Description must include proof that the applicant has the services of a qualified person responsible for pharmacovigilance.
The description of the marketing authorization holder’s pharmacovigilance system should follow the requirements and format as detailed in Volume 9A of EudraLex.
Risk-management System (Section 1.8.2)
Detailed description of risk-management system must be provided according to Article 8 (ia) of Directive 2001/83/EC. The detailed description of a risk management system should be provided in the form of an EU Risk Management Plan (EU-RMP), as outlined in Volume 9A of EudraLex.
Information related to Clinical Trials (Section 1.9)
The section of 1.9 must be prepared according to Article 8 (ib) of Directive 2001/83/EC for report to the effect that clinical trials carried out outside the European Union meet the ethical requirements of Directive 2001/20/EC should be provided, where applicable.
This section should be provided for all new applications (including extension applications), and other relevant post-authorization regulatory procedures (e.g., variations) for which clinical trial submitted reports.
Information relating to Pediatrics (Section 1.10)
In accordance with Article 7, 8 and 30 of Regulation (EC) No 1901/2006 (‘pediatric regulation’) and Article 23 of Regulation (EC) No 1901/2006 (‘pediatric regulation’) this section is required:
- For all new applications*, for a medicinal product which is not authorized in the EEA
- For applications* for new indications, new pharmaceutical forms and new routes of administration, for authorized medicinal products which are protected either by a supplementary protection certificate, or by a patent which qualifies for the granting of such a certificate.
- For Pediatric Use Marketing Authorization Applications (PUMA)
*except for generic, hybrid, bio-similar and well-established use applications and traditional herbal or homeopathic medicinal products
For Module 1 can be provided:
Responses to Questions in those instances when applicants are advised to include in this section a document which lists the questions with the corresponding narrative text response for each question, and when responses also contain new or updated data/documents related to Modules 3, 4 and/or 5. Such data/documents should be placed in the relevant sections of those Modules.
Additional Data. This section is required based on authorization procedure. Additional data may need to be provided as part of a national, decentralized or mutual recognition application. If such data related to Modules 2, 3, 4 and/or 5, the documents should also be placed in the relevant sections of those Modules. Member State specific requirements for additional data can be found on the Website of the European Commission.
CTD-Module 2
The general introduction to the medicinal product provided in Module 2 section of CTD dossier, which is harmonized for all regions (The International Council for Harmonization of Technical Requirements for Pharmaceuticals for Human Use (ICH)). This module presented by summary documents for each upcoming modules: quality data, non-clinical and clinical study reports.
CTD-Module 3
Module 3 was identified by ICH as the Quality Module. Thus, “Quality” became the global term for CMC (chemistry, manufacturing and controls). CMC should not be confused with the elements of quality control, quality assurance, SOPs (standard operating procedures), internal company documents (specifications, batch records, etc.)
Module 3 section also harmonized for all regions with providing information of chemical-pharmaceutical and biological information for chemical active substances and biological medicinal products.
Throughout development and after a product is approved by health authorities, the chemistry, manufacturing and control (CMC) aspects continue to evolve and change.
Pharmaceutical Quality = CMC
CHEMISTRY
Discovery of new chemical entities/molecules, purification of active ingredients (drug substance), analytical testing of raw materials and finished products.
MANUFACTURING
Lab scale manufacturing of drug substances and drug products, manufacture of clinical supplies for clinical studies, scaling up to commercial batch size, commercial product.
AND CONTROLS
Develop appropriate specifications/controls for drug substance and drug product to ensure safety, efficacy and quality.
All regions require CMC/Pharmaceutical Quality dossier
- To initiate and conduct clinical trials,
- To submit a new marketing authorization dossier (MAA, NDA, BLA, etc.)
- To meet the regulatory requirements for the life cycle management and post approval changes of the product
This module includes the whole package of documents regarding drug substance (DS) and drug product (DP).
Drug Substance (DS)
Drug substance section is presented by its description, including physical, chemical, or biological characteristics, the name and address of DS manufacturer, the acceptable limits and analytical methods used to assure the identity, strength, quality, and purity of the drug substance.
Information to support the stability of the drug substance during the toxicologic studies and the proposed clinical study is also included.
DRUG PRODUCT (DP)
A list of all components, which may include reasonable alternatives for inactive compounds, used in the manufacture of the drug product, including both those components intended to appear in the drug product and those which may not appear, but which are used in the manufacturing process are described in this section.
Below is listed key information of drug product which is required to be included in regulatory dossier:
- The quantitative composition of the (investigational or commercial) drug product, including any reasonable variations.
- The name and address of the drug product manufacturer.
- Description of the manufacturing and packaging procedures.
- The acceptable limits and analytical methods used to assure the identity, strength, quality, and purity of the drug product.
CTD-Module 4
Nonclinical Study Reports
The relevant section the appropriate location for individual-animal data is in the study report in the Common Technical Document for applications that will be submitted to Regulatory Authorities. Module 4 is harmonized for US and EU based on ICH principals, and contains all necessary sections and sub-sections for study reports. References to ICH guidelines Common Technical Document (CTD).
CTD-Module 5
Clinical Study Reports
Module 5 section this is the structure and content of clinical study reports. This part of CTD presented human/clinical study reports, other clinical data, and references within a Common Technical Document (CTD) for registration of a pharmaceutical product for human use. These elements should facilitate the preparation and review of a marketing application. Module 5 is harmonized for US and EU based on ICH principals, and contains all necessary sections and sub-sections for study reports. References to EU guidelines Common Technical Document (CTD).
Need help to prepare a regulatory dossier? View and consult freelance regulatory writers on Kolabtree.
Additional References
- https://www.ich.org/page/ctd
- https://www.fda.gov/media/128163/download
- https://www.fda.gov/media/135573/download
- https://ec.europa.eu/health/sites/default/files/files/eudralex/vol-2/b/update_200805/ctd_05-2008_en.pdf
- https://ec.europa.eu/health/sites/default/files/files/eudralex/vol-1/dir_2001_83_consol_2012/dir_2001_83_cons_2012_en.pdf
- https://eur-lex.europa.eu/legal-content/EN/TXT/PDF/?uri=CELEX:02001L0083-20121116&from=DE
- https://eur-lex.europa.eu/legal-content/EN/TXT/PDF/?uri=CELEX:02004R0726-20130605&from=EN
- https://ec.europa.eu/health/sites/default/files/files/eudralex/vol-1/reg_2000_141_cons-2009-07/reg_2000_141_cons-2009-07_en.pdf
- https://www.ema.europa.eu/en/human-regulatory/research-development/clinical-trials-human-medicines
- https://eur-lex.europa.eu/legal-content/EN/TXT/?uri=celex%3A32006R1901
- https://clinicaltrials.gov/ct2/home
Kolabtree helps businesses worldwide hire freelance scientists and industry experts on demand. Our freelancers have helped companies publish research papers, develop products, analyze data, and more. It only takes a minute to tell us what you need done and get quotes from experts for free.

Unlock Corporate Benefits
• Secure Payment Assistance
• Onboarding Support
• Dedicated Account Manager
Sign up with your professional email to avail special advances offered against purchase orders, seamless multi-channel payments, and extended support for agreements.


