



An exhaustive guide to conformité réglementaire for IVD manufacturers, written by Sundeep Agarwalexpérimenté Consultant de l'IVDR.
Qu'est-ce que l'IVDR ?
The European Commission’s (EC) In Vitro Diagnostic Regulation (EU IVDR 2017/746) is a ‘legislative framework’ and a way forward towards global IVD safety, which assures that only reliable and effective IVDs are in the market. The European Commission is trying its best to make the soins de santé system safer and error free in terms of diagnosis or outcomes.
Les dispositifs médicaux de diagnostic in vitro (IVDD), 98/79/CE était une directive tandis que l'IVDR est une législation (règlement) applicable à tous les opérateurs économiques (OE), c'est-à-dire les fabricants, les importateurs, les utilisateurs, les organismes notifiés et les autorités nationales dans l'Espace économique européen (EEE) et les fabricants et fournisseurs non européens qui placent ou prévoient de distribuer des DIV sur le marché européen.
L'IVDR comprend 113 articles (10 chapitres) et 15 annexes, contre 24 articles et 10 annexes pour l'IVDD. Il ne fait aucun doute que l'IVDR est un long texte réglementaire et qu'il est très strict, mais le point positif est qu'il est plus transparent en ce qui concerne les changements et les exigences réglementaires.
Elle met l'accent sur une approche basée sur le cycle de vie. Elle sera appliquée à partir du 26 mai 2022 et les opérateurs économiques (y compris les fabricants hors UE) doivent se préparer de manière proactive à la planification et à la mise en œuvre de cette directive. Chaque partie prenante au processus sera désormais également responsable du marché du diagnostic in vitro de l'Espace économique européen (EEE).
- La première et principale chose qu'une organisation doit faire est d'organiser un programme de formation (en ligne ou sur place, selon le cas) sur l'IVDR de l'UE, afin que tous les membres de l'organisation soient conscients des changements nécessaires.
- Une communication officielle doit être suivie par, à tous les fournisseurs, sous-traitants, ou prestataires de services sur le processus et leurs obligations.
- Réaliser une évaluation des écarts pour vérifier la disponibilité de leurs ressources, une équipe compétente pour mettre à jour la documentation technique requise dans le cadre de l'EU IVDR. Être certifié ISO 13485 : 2016 serait un avantage supplémentaire pour établir la conformité.
- Il est conseillé (si nécessaire) d'engager un expert en la matière ou un consultant externe dès le tout début de la transition, car "un point à temps en vaut neuf".
- Ce blog fournira un aperçu détaillé et des conseils pratiques pour se conformer aux attentes des organismes notifiés et des autorités compétentes telles que décrites dans les différents articles et annexes du règlement européen IVDR 2017/746.
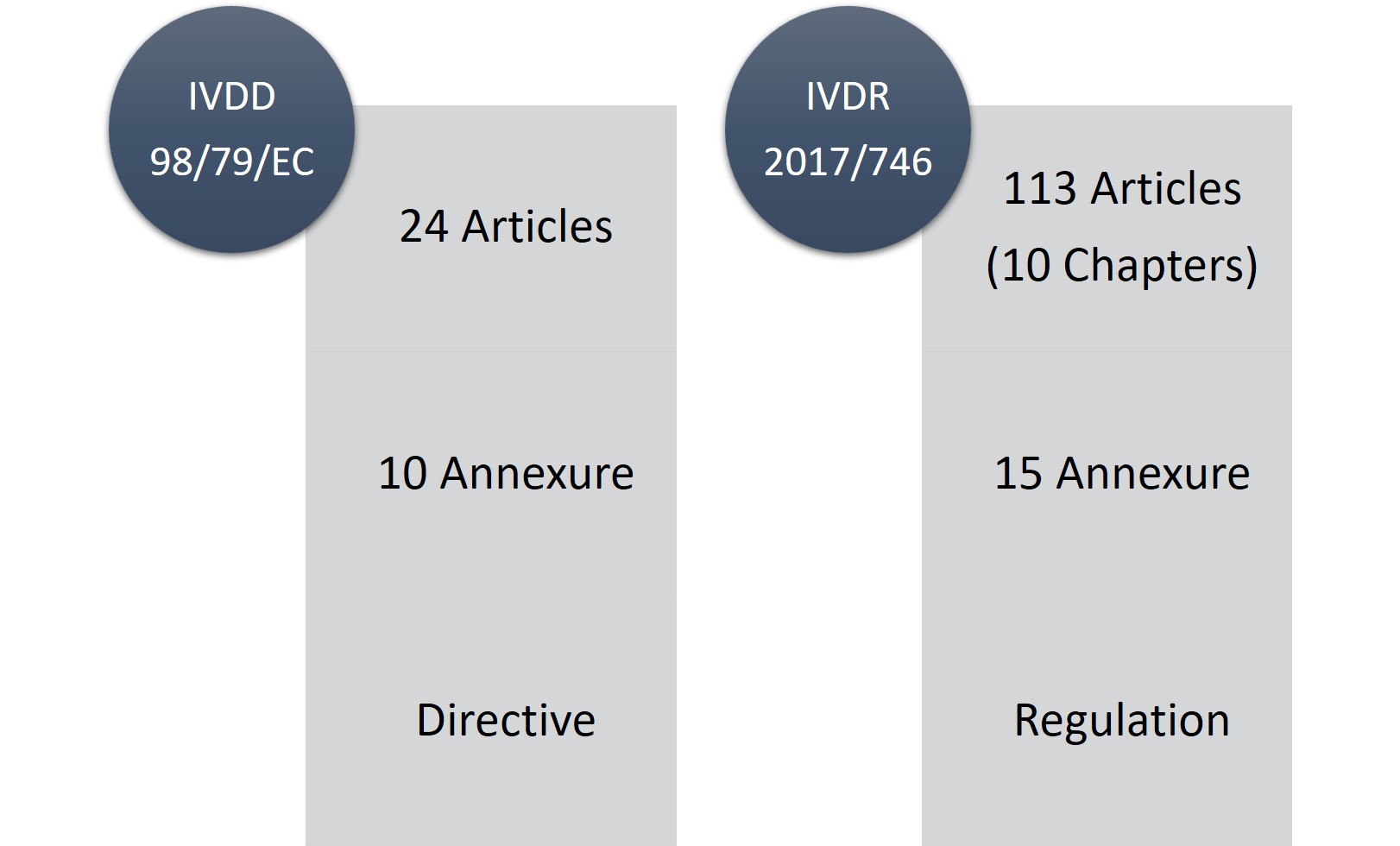
Figure1 : IVDD vs. IVDR
1. Se préparer à la conformité à l'IVDR et aux changements commerciaux
La principale décision commerciale pour une organisation est de déterminer si elle souhaite continuer à placer ses DIV dans l'Espace économique européen (EEE). Si la réponse est "oui", il faut alors obtenir le plus tôt possible d'un ON des estimations (coût), des délais, la portée de l'audit, le code du produit, etc. Le passage d'une directive à un règlement exige une conformité obligatoire et une documentation technique solide pour établir la sécurité et l'efficacité et obtenir la certification CE. L'IVDR s'appuie beaucoup plus sur les preuves cliniques. c'est-à-dire la validité scientifique, les performances analytiques et les performances cliniques. pour établir la sécurité et l'efficacité.
L'implication d'un organisme notifié (ON) dans le processus de certification CE sera une caractéristique importante du règlement. Cela indique également un investissement supplémentaire pour l'opérateur économique qui peut indirectement augmenter le coût du produit.
Une nomination d'un "Personne responsable de la conformité réglementaire (PRRC)". in accordance with Article 15 of EU IVDR 2017/746 is now mandatory; who shall assure the conformity of QMS, declaration of conformity, technical documentation, post market surveillance and reporting of adverse events are in compliance to EU IVDR.Manufacturers should ensure that the entire transition (including new certification application) is completed before the expiry of their existing IVDD Certificate or Self-certified Declaration of conformity. Certificates issued by notified bodies in accordance with IVDD 98/79/EC from 25 May 2017 shall become invalid after 27 May 2024. Be aware of the new timeline for application as per the EC official press release [1] dtd.20th Décembre 2022.
2. Compréhension claire de la classification
Reconsidérez la nouvelle règle de classification de l'annexe VIII de l'IVDR et vérifiez si elle a affecté votre classification précédente.
Il est essentiel de procéder à une classification correcte avant de se préparer au processus de certification CE. Si nous ne sommes pas en mesure de le faire, le parcours de conformité ne sera pas clair et retardera ou invalidera nos efforts pour nous conformer aux exigences de l'IVDR.
L'IVDR est un approche fondée sur le risque pour classer les dispositifs avec des contrôles accrus de l'organisme notifié et de l'autorité compétente. Le règlement identifie quatre classes de risque: Classe A (risque le plus faible), Classe B, Classe C, et Classe D (risque le plus élevé) tandis que l'annexe VIII définit sept règles de classification pour classer correctement les produits. Une caractéristique unique de l'IVDR est que les logiciels sont également classés en vertu de la règle d'application 1.4 de l'annexe VIII, qui stipule que "les logiciels qui commandent un dispositif ou influencent l'utilisation d'un dispositif doivent appartenir à la même classe que le dispositif. Si le logiciel est indépendant de tout autre dispositif, il doit être classé en tant que tel.[2]". Cela indique le champ d'application du logiciel à réglementer dans le cadre de l'IVDR. Et le fabricant doit également effectuer la vérification et la validation du logiciel (Annexe II, 6.4) en conséquence.
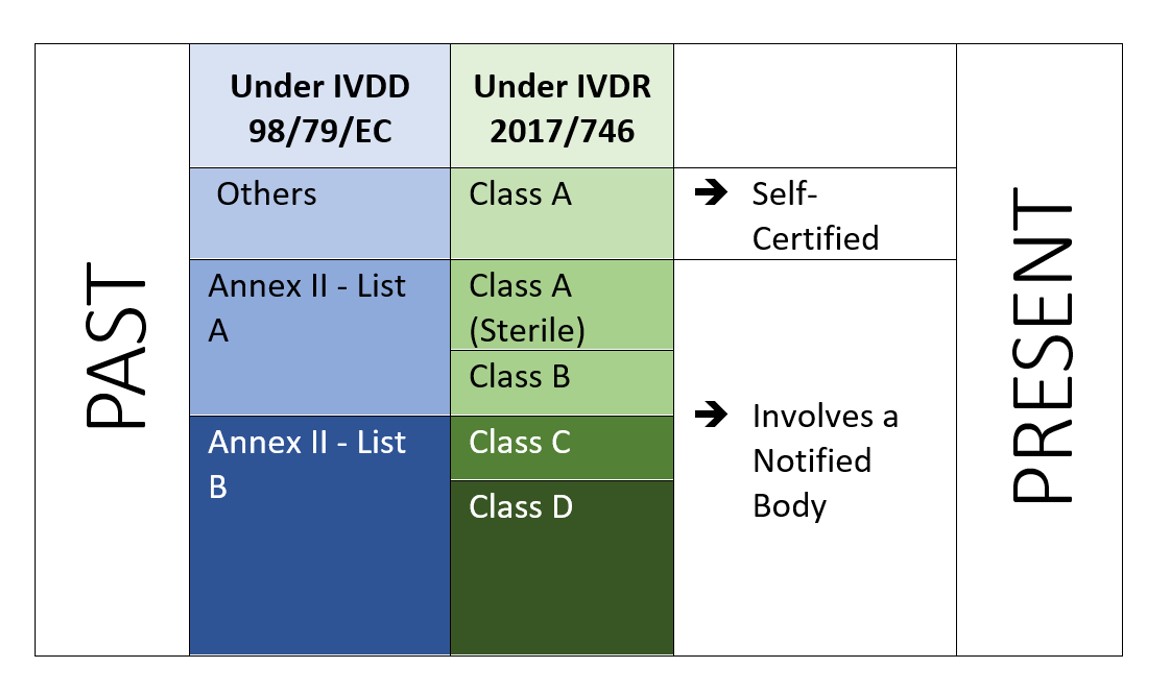
Figure 2 : Classification basée sur le risque dans le cadre de l'IVDR 2107/746
3. Participation de l'organisme notifié
Le rôle d'un organisme notifié (ON) serait l'un des éléments essentiels et, par conséquent, un plus grand nombre de fabricants devraient désormais être audités et certifiés par un organisme notifié par rapport à la méthode traditionnelle d'"auto-certification".Les opérateurs économiques doivent choisir avec soin la voie de l'évaluation de la conformité (annexe IX, X, XI de l'IVDR de l'UE).[3]).
L'IVDR n'exige pas seulement un investissement supplémentaire, mais il faut aussi s'assurer que la documentation technique et le système de gestion de la qualité répondent aux nouvelles exigences de l'IVDR. Dans le cadre de l'IVDD, la plupart des DIV sont auto-certifiés (92%) et ne nécessitent pas l'intervention d'un organisme notifié (sauf 8% de l'ensemble des DIV mis sur le marché [4]). Alors que dans le cadre du nouvel IVDR, le scénario n'est pas le même.
Selon une étude “The impact of the new European IVD-classification rules on the notified body involvement” by National Institute for Santé publique and the Environment, Bilthoven (Netherlands) RIVM Letter report 2018-0082, A. van Drongelen et al., nearly 85% of all IVDs will be requiring Notified Body involvement, leaving only 15% of IVDs eligible for self-certification[5].
Cela signifie également que les fabricants de produits de diagnostic in vitro (DIV) vont devoir se conformer au nouveau processus de classification et de certification. En outre, en fonction de l'utilisation prévue des dispositifs et de la classe de risque, les fabricants doivent identifier un ON désigné qui pourrait être en mesure de les auditer et de faire certifier leurs produits. Les DIV à haut risque (classe D) nécessiteraient un laboratoire de référence de l'UE ou des groupes d'experts pour vérifier la déclaration de performance en plus de l'implication d'un organisme notifié (ON) ou d'une autorité compétente (AC). Actuellement, seuls six organismes notifiés sont désignés dans le cadre de l'IVDR de l'UE. N'attendez pas pour commencer votre processus de demande afin d'éviter des retards inattendus dus à l'indisponibilité d'un organisme notifié.

Figure 3 : Liste des organismes notifiés désignés dans le cadre de l'IVDR [6].
4. Mise en place du système de gestion de la qualité (SGQ)
Les fabricants de DIV sont censés établir un système de gestion de la qualité (SGQ) robuste et fiable dans leurs locaux. Il s'agit d'une obligation générale d'un fabricant en vertu de l'article 10 de l'IVDR.. Le système de gestion de la qualité est une exigence essentielle parmi d'autres, sans laquelle un fabricant ne pourra pas être agréé.
QMS is to ensure that manufacturing, change control, customer complaints, resource management, supplier &sub-contractors’ controls and validation, performance evaluation, quality test, UDI Labelling, Post market surveillance etc. are according to approved QMS and Post Market Surveillance (PMS) plans.
Le CRRP doit s'assurer que le fabricant a satisfait aux exigences de l'article 10 pour "autocertifier" (délivrance d'une déclaration de conformité conformément à l'annexe IV) les DIV de classe A lorsqu'un organisme notifié (ON) n'est pas requis dans le processus.
5. Se préparer aux perturbations de la chaîne d'approvisionnement
Throughout the world, manufacturer depends largely on their supply chain and raw material to produce and deliver IVDs that are safe, accurate, and effective forthe intended use. Hence regulatory and quality concerns are also evolving to a higher level when it comes to the suppliers and sub-contractors’ controls. Manufacturer are therefore expected to proactively communicate the supply chain about their obligations and responsibilities of the suppliers and subcontractors. Legal manufacturer shall demonstrate adequate supplier control and monitoring, assure the supply chain is in compliance to the regulatory aspects of IVDR, reconsider the need for data integrity and quality of supplier data, implement robust supplier risk management and performance monitoring and periodically audit the supplier based on the associated risk to the finished products. Les régulateurs et les organismes notifiés insistent sur le fait que les fabricants légaux doivent clairement documenter le niveau de contrôle des fournisseurs et démontrer, preuves à l'appui, qu'ils sont en mesure d'atténuer le risque lié au produit ou au service fourni par le fournisseur.
6. Assurer la préparation aux audits et aux inspections
Selon l'article 88 de l'IVDR, Activités de surveillance du marché, les autorités compétentes doivent effectuer des inspections annoncées (inopinées) dans les locaux des opérateurs économiques ainsi que des fournisseurs et/ou des sous-traitants et, si nécessaire, dans les installations des utilisateurs professionnels. Le fabricant doit inclure des informations sur l'identification de tous les sites, y compris les fournisseurs et les sous-traitants, où les activités de fabrication sont réalisées dans la documentation technique des informations sur la conception et la fabrication. Les organismes notifiés (ON) qui effectuent l'audit du SMQ doivent identifier les liens entre les différents sites de fabrication et leurs fournisseurs et/ou sous-traitants ainsi que la répartition des responsabilités entre eux. Ces informations seront prises en compte si l'ON souhaite spécifiquement auditer l'un de ces fournisseurs ou sous-traitants ou les deux. Les locaux des fournisseurs du fabricant, lorsqu'ils sont considérés comme affectant de manière significative la conformité des dispositifs finis, seront essentiellement audités par l'ON (en particulier lorsque le fabricant ne peut pas démontrer un contrôle suffisant sur ses fournisseurs).
7. Plan pour gérer les audits inopinés
Dans le cadre de la surveillance post-certification, l'ON procède à des audits inopinés sur place des fabricants et de leurs sous-traitants ou fournisseurs effectuant des tests de produits et à la surveillance de la conformité à toutes les conditions liant les fabricants et associées aux décisions de certification, telles que les mises à jour des données cliniques à des intervalles définis.En outre, tL'organisme notifié effectue, au moins une fois tous les cinq ans, des audits inopinés sur le site du fabricant et, le cas échéant, sur le site des fournisseurs et/ou des sous-traitants du fabricant, qui peuvent être combinés avec l'évaluation de surveillance périodique.
8. Renforcer les activités de surveillance après la mise sur le marché
Il est fortement recommandé aux fabricants de renforcer leurs exigences en matière de surveillance post-commercialisation et de développer un mécanisme de coordination entre les États membres de l'UE en matière de vigilance et de surveillance du marché. Dans le cadre de l'évaluation de la surveillance applicable aux dispositifs de classe C et de classe D (annexe IX), l'organisme notifié effectue périodiquement, au moins une fois tous les 12 mois, des audits et des évaluations appropriés. Cela comprend des audits dans les locaux du fabricant et des fournisseurs et/ou sous-traitants, le cas échéant. Le fabricant doit essentiellement élaborer une procédure pour l'enregistrement et la notification des incidents et des actions correctives de sécurité sur le terrain (FSCA).
9. Identifiant unique du dispositif (UDI) & EUDAMED
Le fabricant doit mettre en place un système d'identification unique (UDI) pour identifier et faciliter la traçabilité des dispositifs. On peut se référer à une liste d'entités émettrices accréditées (IE) telles que GS1, HIBCC, ICCBBA, IFA GmbH pour gérer un système d'attribution d'UDI. Actuellement, les IE mentionnées sont valables à partir du 27th juin 2019, mais il sera sage de confirmer leur validité avant de prendre une décision finale quant à leur mise en œuvre.
La base de données européenne sur les dispositifs médicaux (EUDAMED) fournira un aperçu de tous les dispositifs médicaux disponibles dans l'Union européenne. Elle se compose de six modules relatifs à :
- Enregistrement de l'acteur,
- Identification unique des dispositifs (UDI) et enregistrement des dispositifs,
- Organismes notifiés et certificats,
- Investigations cliniques et études de performance,
- Vigilance et surveillance post-marché, et
- Surveillance du marché.
Afin d'assurer une meilleure transparence grâce à un EUDAMED complet, une partie des informations des opérateurs économiques sera accessible au public. Les informations confidentielles ne seront accessibles qu'aux opérateurs économiques, aux promoteurs, aux autorités notifiées et aux autorités compétentes des États membres de l'UE.
10. Exigences pour les "dispositifs internes".
Health institution developing ‘in-house devices’ (or ‘laboratory-developed tests’) which are meant to be used by the same health institution shall not be marketed or sold to other legal entity. Such devices may be used for the diagnosis and treatment, especially for rare diseases. The institution is expected to comply with only the requirement of Annex I of IVDR (general safety and performance requirements), and exempted from rest of the regulation until 26 May 2024; provided the health institution meets a number of conditions set out in Article 5(5) of the Regulation and has an appropriate quality management system, which complies to the international standard setting out the quality and competence requirements for medical laboratories (EN ISO 15189) or other national provisions, and is able to justify that target patient group’s specific needs cannot appropriately be met by an equivalent device available on the market.
Références
[1] Communiqué de presse officiel de la CE, daté du 20 décembre.th Déc 2021, mise en place progressive du règlement sur les dispositifs médicaux de diagnostic in vitro. Peut être consulté à l'adresse suivante https://ec.europa.eu/commission/presscorner/detail/en/IP_21_6965 [2]RÈGLEMENT (UE) 2017/746 DU PARLEMENT EUROPÉEN ET DU CONSEIL du 5 avril 2017 relatif aux dispositifs médicaux de diagnostic in vitro et abrogeant la directive 98/79/CE et la décision 2010/227/UE de la Commission ; ANNEXE. VIII RÈGLES DE CLASSIFICATION, 1. RÈGLES DE MISE EN ŒUVRE Point 1.4 Page 304 [3]Règlement (UE) 2017/746 du Parlement européen et du Conseil du 5 avril 2017 relatif aux dispositifs médicaux de diagnostic in vitro et abrogeant la directive 98/79/CE et la décision 2010/227/UE de la Commission.ANNEXE IX Évaluation de la conformité sur la base d'un système de gestion de la qualité et de l'évaluation de la documentation technique, Page 306, ANNEXE X Évaluation de la conformité sur la base d'un examen de type, Page 314, ANNEXE XI Évaluation de la conformité sur la base de l'assurance de la qualité de la production, Page 317
[4] Communiqué de presse dtd. 14 octobre 2021, Bruxelles ; Santé publique : La Commission propose une mise en œuvre progressive du nouveau règlement sur les dispositifs médicaux de diagnostic in vitro. [5]L'impact des nouvelles règles européennes de classification des DIV sur la participation de l'organisme notifié : une étude sur les DIV enregistrés aux Pays-Bas ; van Drongelen A, de Bruijn A, Pennings J, van der Maaden T 32 p en anglais 2018, RIVM letter report 2018-0082 [6] La liste ci-dessus est basée sur les données consultées en date du 5 mars 2021. 5 mars 2021, pour les dernières mises à jour de la liste, vous pouvez accéder au site officiel de la CE à l'adresse suivante https://ec.europa.eu/growth/tools-databases/nando/index.cfm?fuseaction=directive.notifiedbody&dir_id=35Kolabtree helps businesses worldwide hire freelance scientists and industry experts on demand. Our freelancers have helped companies publish research papers, develop products, analyze data, and more. It only takes a minute to tell us what you need done and get quotes from experts for free.

Unlock Corporate Benefits
• Secure Payment Assistance
• Onboarding Support
• Dedicated Account Manager
Sign up with your professional email to avail special advances offered against purchase orders, seamless multi-channel payments, and extended support for agreements.


