



Shrinidh Joshi, experto en dispositivos médicos on Kolabtree, provides a comprehensive guide to dispositivo médico design, design controls, validation & verification, regulatory requirements and risk management.
En el artículo anterior, we took a look at the overview of the dispositivo médico development process from the ideation to the discovery phase. In this article, we will focus more on medical device design, design controls, and compliance.
Diseño de dispositivos médicos: Normativa IEC e ISO y su cumplimiento
A estas alturas ya sabe que, para entrar en el mercado, su producto sanitario debe cumplir ciertos requisitos y normas reglamentarias. Las normas de productos sanitarios, como la Comisión Electrotécnica Internacional (CEI) o la Organización Internacional de Normalización (ISO), permiten a los fabricantes de productos sanitarios, diseñadores, laboratorios y todos los demás proveedores de servicios de desarrollo de productos sanitarios, como los CDMO, inspeccionar, evaluar y mantener sus dispositivos y equipos de acuerdo con determinadas normas de calidad y usabilidad.
La CEI publicó la primera norma de este tipo sobre productos sanitarios en 1970, IEC 60601-1. IEC 60601-1, Equipos electromédicos - Parte 1: Esta es la norma reconocida internacionalmente que aborda los requisitos generales para los equipos y dispositivos electromédicos que cubren las normas de seguridad básica y rendimiento esencial [4].
El documento IEC 60601-1 ha sido revisado periódicamente para mantenerse en línea con los últimos desarrollos médicos y avances tecnológicos en el campo de los dispositivos médicos. El cambio más reciente se produjo en 2012 (Enmienda 1 a la norma IEC 60601-1). Esta norma revisada contiene los requisitos para la consideración del factor humano, la evaluación del rendimiento esencial de los productos sanitarios, la usabilidad y los comandos. También incluye el software como dispositivo médico y especifica su adopción de un ciclo de vida de desarrollo formal. En el ámbito de la norma IEC 60601-1 revisada también se incluyen especificaciones técnicas más nuevas y revisadas sobre riesgos (tanto eléctricos como mecánicos), requisitos de etiquetado de productos sanitarios (incluidas nuevas normas de etiquetado) y documentación.
Diseño de dispositivos médicos: Normas ISO
La Organización Internacional de Normalización también dispone de especificaciones para las normas de los productos sanitarios. ISO 13485 y ISO 14971 son normas ampliamente utilizadas en todo el mundo para la gestión de la calidad de los productos sanitarios. Aparte de estas normas internacionales, algunas normas son específicas de cada región y todas ellas se adoptan de las normas internacionales con pocas modificaciones y limitaciones.
Si una empresa de productos sanitarios fabrica o vende productos sanitarios en Estados Unidos, el producto sanitario estará regulado por la FDA. El American National Standards Institute (ANSI) es el representante de las normas ISO en Estados Unidos.
Hay otras dos organizaciones similares: la Asociación para el Avance de la Instrumentación Médica (AAMI) y la Sociedad Americana para la Calidad (ASQ), que define las normas para Estados Unidos.
Si una empresa de productos sanitarios ha diseñado un producto teniendo en cuenta las normas ISO, también existe la posibilidad de que la FDA no apruebe el producto. Como la FDA tiene su propio conjunto de procedimientos para la gestión de riesgos derivados de las normas internacionales y regionales, que incluye:
(norma internacional.)
- ANSI/AAMI/ISO 14971:2007 (R2010), Productos sanitarios - Aplicación de la gestión de riesgos a los productos sanitarios (Una norma regional con adiciones y modificaciones de la referida norma internacional). [5].
En el caso de la norma de gestión de la calidad, no sigue la versión internacional o regional de la norma ISO 13485. Esto se debe a que la FDA tiene directrices diferentes para la gestión de la calidad en los productos sanitarios para el mercado estadounidense.
Mientras que, si la empresa de productos sanitarios está considerando la Unión Europea, el Comité Europeo de Normalización (CEN) es la normalización adoptada de la ISO y el Comité Europeo de Normalización Electrotécnica (CENELEC) es la norma regional inspirada en la CEI.
El CEN está un poco modificado según los requisitos de la ISO y se escribe con el prefijo "EN". Por ejemplo
- EN ISO 13485:2012, Productos sanitarios - Sistemas de gestión de la calidad - Requisitos para fines reglamentarios.
- EN ISO 14971:2012, Productos sanitarios - Aplicación de la gestión de riesgos a los productos sanitarios
Los miembros nacionales adoptan estas normas de la UE añadiendo su prefijo. En el caso de Suiza, Swiss Standards publica normas con "SN" como prefijo, como SN EN ISO 13485:2012 y SN EN ISO 14971:2012.
En el caso de Canadá, la Autoridad Canadiense de Normalización (CSA) es la organización que representa a la ISO.
Normativa sobre productos sanitarios y control del diseño
Los fabricantes de dispositivos médicos deben seguir las directrices de control de diseño, ya que los organismos reguladores como la FDA, la Comisión Europea, el Ministerio de Sanidad de Canadá y otros quieren asegurarse de que los dispositivos médicos son seguros para los usuarios potenciales antes de que los fabricantes empiecen a comercializarlos. Como he mencionado en la sección anterior, la FDA no sigue la norma ISO 13485, ya que tiene diferentes requisitos para la gestión de la calidad. Los controles de diseño se definen en FDA 21 CFR 820.30 que tiene una intención similar a la sección 7.3 Diseño y desarrollo descrita en las directrices de la norma ISO 13485. Además, la FDA incorpora los requisitos de las buenas prácticas de fabricación actuales (cGMP) en la regulación del sistema de calidad para seguir las buenas prácticas de calidad para los diseños de dispositivos médicos [6].
La normativa proporciona un marco para aplicar el control del diseño a una amplia variedad de dispositivos. El marco ofrece flexibilidad tanto para el cumplimiento de la normativa como para el proceso interno de diseño y desarrollo.
Para llevar a cabo con éxito el control del diseño de los dispositivos médicos, se necesitan profesionales con formación tanto técnica como no técnica, como administración de empresas, ciencias de la vida, ingeniería, informática y artes.
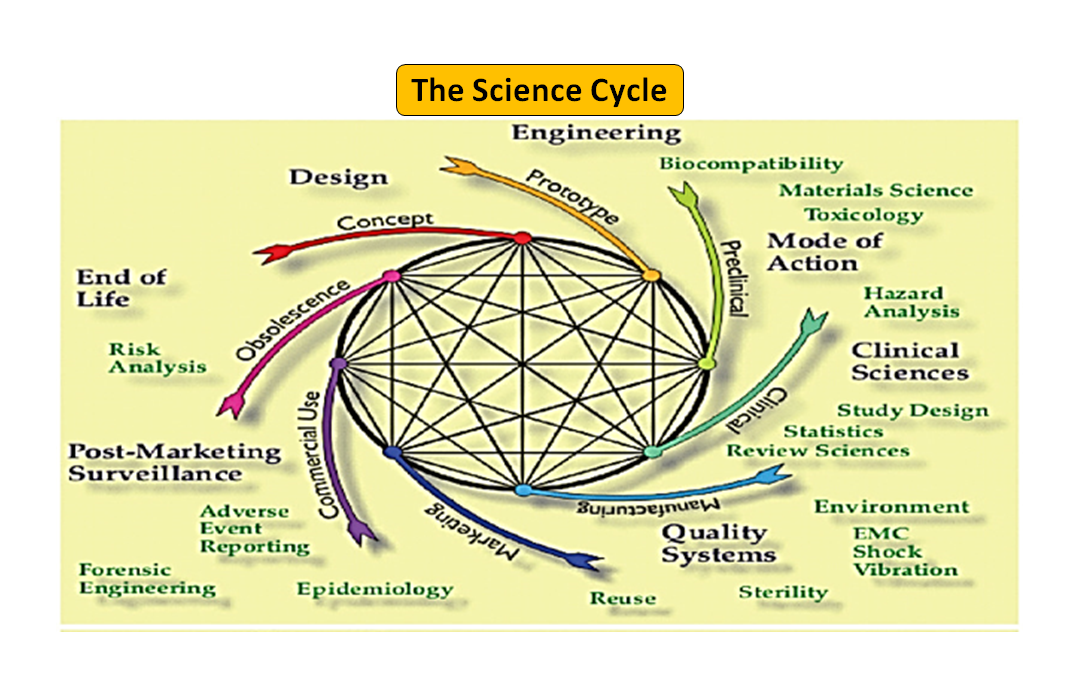
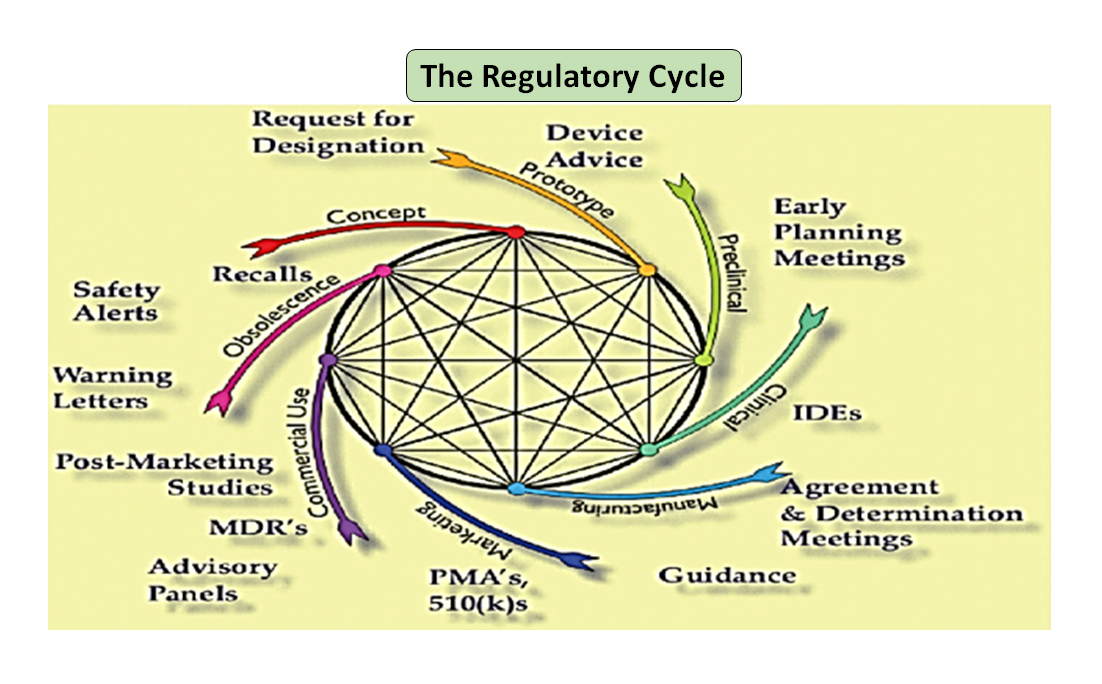
Figura 1: Ciclo de vida total del producto. El ciclo científico y el ciclo normativo (Adaptado de [7]).
Cabe destacar que el ciclo de vida de los dispositivos médicos, desde la innovación hasta la aprobación reglamentaria y la comercialización, es una serie de pasos interconectados que impulsan el desarrollo del dispositivo (véase la Figura 1: Ciclo total del producto). Al principio, los prototipos diseñados por sus ingenieros se someten a pruebas de banco para optimizar el diseño, y se comprueba la biocompatibilidad, los extraíbles, los lixiviables, la flexibilidad o la resistencia general del dispositivo. La función del asesor en materia de reglamentación de su empresa es buscar en la base de datos de reglamentación para sugerirle un documento de orientación que pueda ayudarle a determinar si su producto será regulado como producto sanitario o no. El uso previsto de su producto sanitario y su modo de funcionamiento o acción le guiarán en el diseño del producto y también decidirán su vía de regulación, ya sea 510(k), PMA, De Novo, Pre-sub, IDE, HDE, archivos maestros, etc.
Como se ilustra en la figura 1, tanto los procesos científicos como los reglamentarios se entrelazan a lo largo del ciclo de vida del producto. Al igual que las diferentes partes del ciclo de vida de la ciencia están interconectadas, la ciencia y los requisitos reglamentarios están entrelazados, cada uno informando y determinando al otro. Existe la oportunidad de establecer conexiones, tanto en la FDA como en los fabricantes, para que las partes del ciclo de vida no corran el riesgo de ser consideradas de forma aislada. Por ejemplo, no es raro que se revise una solicitud previa a la comercialización sin tener en cuenta la experiencia posterior a la comercialización de productos similares.
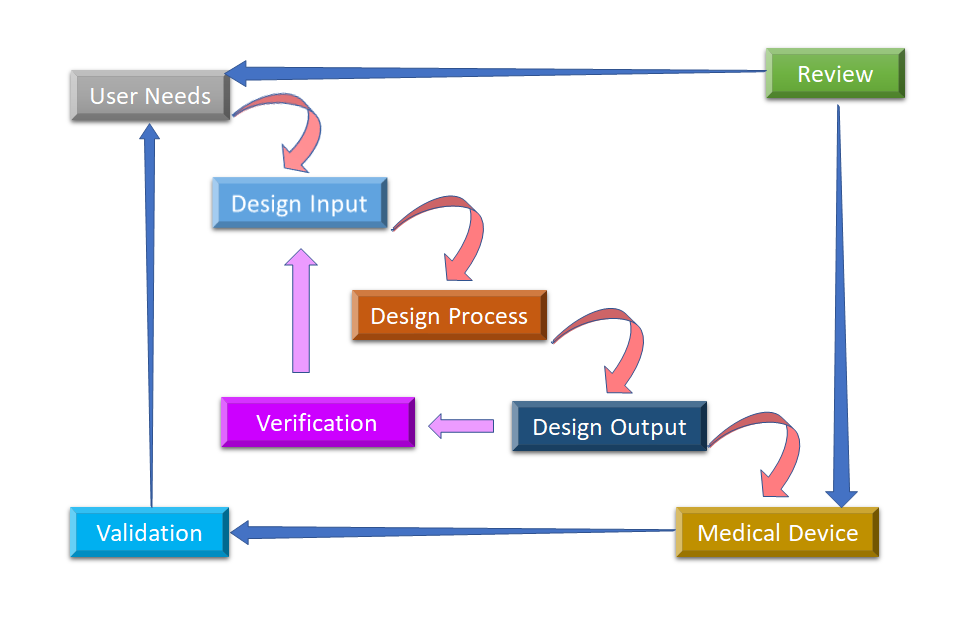
Figura 2: Proceso de diseño del flujo de agua para el control del diseño de dispositivos médicos (Adaptado de [8]).
La fase inicial de la que parte el control del diseño es el desarrollo y la aprobación de la entrada del diseño, que consiste en el diseño del dispositivo y los procesos de fabricación que se llevarán a cabo en la fase de producción. El control del diseño es un enfoque holístico y no termina con la transferencia del diseño a la fase de producción, una vez que el diseño está finalizado. También influye en los procesos de fabricación en función de los cambios en la fase de diseño o incluso de los comentarios posteriores a la producción. Es un proceso continuo para desarrollar un producto que sea utilizable para un usuario y, por lo tanto, para el producto mejorado, considera los cambios revolucionarios a partir de los patrones de uso, así como el análisis de los productos fallidos. En la figura 2 se puede ver cómo se realiza el control del diseño en el proceso de diseño en cascada.
- Necesidades del usuario:- Los requisitos se definen teniendo en cuenta la necesidad del mercado y el dispositivo se diseña para satisfacer esa necesidad. Tras una serie de evoluciones, se finaliza el diseño del dispositivo médico y se traslada a producción para su fabricación. En cada etapa de este proceso es necesario obtener información de retorno.
- Entrada de diseño: Se trata de un proceso iterativo. Cuando una organización decide abordar una necesidad concreta, revisa y comprueba la aceptabilidad del diseño derivado de la necesidad. En ese momento, se inicia el proceso iterativo de conversión de los requisitos en el diseño del dispositivo.
- Proceso de diseño: Estas entradas de diseño se convierten en salidas de diseño al convertir esos requisitos en especificaciones de alto nivel (que son salidas de diseño).
- Diseño de salida: El proceso de verificación confirma si las especificaciones satisfacen los requisitos o no. Y el resultado se convierte en la entrada para revisar los requisitos y este proceso continúa hasta que la salida del diseño se alinea con la entrada del diseño.
- Dispositivo médico: Once the final design is ready, it is transmitted to the production facility for mass manufacturing. Design control regulation mandates Design History File (DHF), which illustrates the linkages and relationships between all the Design Controls and help to trace all changes throughout the entire desarrollo de productos proceso.
Las empresas de productos sanitarios pueden adoptar un enfoque basado en el papel o en el software, especialmente desarrollado para el control del diseño; los archivos del historial de diseño deben ser rastreables, así como accesibles para todos los miembros del equipo.
El siguiente diagrama de flujo muestra un caso práctico de control de diseño de dispositivos médicos.

Diseño de dispositivos médicos: Por qué es importante la trazabilidad
Currently in the realm of the medical devices industry, it is an ideal practice to develop a traceability matrix that can illustrate the links and relations between user needs, design inputs and outputs, design verification and validation. When you are in the early phase for your device development you can maintain the device traceability using a spreadsheet or document version but as you move forward, its good idea to use cloud-based project management and document sharing platforms such as Microsoft Teams, Asana, Trello or whichever platform is suitable for your organization. The goal is as your project progresses you need to find an option which can save time because the old-school method of maintaining a traceability matrix might consume a lot of your time which you should rather be focusing on design verification and validation.
Una matriz de trazabilidad de los controles de diseño es vital para los equipos de desarrollo de productos, y especialmente para los directores de proyectos, porque la trazabilidad muestra la relación y los vínculos entre todos los controles de diseño. ¿Cómo se relacionan las necesidades del usuario con las entradas de diseño? ¿Cómo se relacionan las salidas del diseño con las entradas del diseño? ¿Cómo se relacionan las verificaciones de diseño con las entradas y salidas de diseño? ¿Cómo se relacionan las validaciones de diseño con las necesidades de los usuarios? Una matriz de trazabilidad es una herramienta inestimable para mostrar una visión de alto nivel y el flujo de desarrollo del producto de dispositivos médicos de principio a fin.
Los desarrolladores de productos con mejores prácticas han confiado en la trazabilidad de los controles de diseño durante muchos, muchos años. Y ahora ISO 13485:2016 también hace de la trazabilidad un requisito. Como se cita en la norma ISO 13485:2016, 7.1 Planificación de la realización del producto, 1. c) las actividades requeridas de verificación, validación, seguimiento, medición, inspección y ensayo, manipulación, almacenamiento, distribución y trazabilidad específicas del producto junto con los criterios de aceptación del producto; Y 7.3.2 Planificación del diseño y el desarrollo, 1. e) los métodos para garantizar la trazabilidad de los resultados del diseño y el desarrollo a los insumos del diseño y el desarrollo. [9].
Diseño de dispositivos médicos: Verificación y validación
Todos los productos sanitarios deben cumplir los objetivos de funcionalidad, facilidad de uso y fiabilidad para conseguir una cuota de éxito en el mercado.
Además, las partes interesadas (pacientes, prescriptores, reguladores o usuarios finales) también prestarán atención a la seguridad y eficacia de su dispositivo. Es muy probable que su dispositivo esté diseñado para satisfacer una necesidad insatisfecha que puede ser crítica para la vida, por ejemplo, un ventilador o un dispositivo de diagnóstico que pueda detectar enfermedades cardíacas. Por lo tanto, es crucial realizar pruebas iterativas de su dispositivo con verificación y validación. Estos dos pasos del proceso de diseño tienen como objetivo confirmar que su dispositivo médico se ajusta a los requisitos de los usuarios y que funciona según su uso previsto. En pocas palabras, la verificación y la validación del diseño pueden garantizar que su dispositivo hace realmente lo que se supone que debe hacer. La verificación y la validación del diseño también sirven para garantizar los requisitos reglamentarios, las normas, la calidad del producto y el proceso de fabricación de su producto sanitario. La verificación del diseño puede evaluar si el resultado de su diseño cumple con los requisitos especificados, las especificaciones o los requisitos reglamentarios que se especifican en la entrada del diseño. Por otro lado, la validación del diseño tiene por objeto evaluar si su producto sanitario ofrece beneficios basados en las necesidades de los usuarios finales.
Diseño verificación pregunta: "¿Hemos diseñado bien el dispositivo?"
Diseño validación pregunta: "¿Hemos diseñado el dispositivo adecuado?"
Los productos sanitarios pueden tener diferentes formas tecnológicas, tamaños y niveles de complejidad. La actividad de verificación y validación (V&V) se rige por el entorno normativo y debe seguir las normas internacionales. Las actividades de V&V estandarizadas pueden agilizar el proceso de fabricación, así como mejorar el proceso de aprobación. Además, las pruebas automatizadas, las técnicas de diagnóstico y las herramientas de recopilación de datos pueden mejorar el proceso de V&V [10].
- Validación de productos frente a validación de procesos
- Diseño de dispositivos médicos/validación de productos: - Conforme a las necesidades del usuario y del paciente, es decir, ¿funciona bien el dispositivo?
- Validación del proceso:- El proceso de fabricación cumple con las especificaciones predeterminadas.
Hay que recordar que la validación del diseño/producto ≠ la validación del proceso. Las agencias reguladoras exigen tanto la validación del diseño/producto como la validación del proceso de forma individual, por lo que ambas deben tenerse en cuenta por igual durante la presentación reglamentaria.
¿En qué momento del proceso de desarrollo hay que pensar en la validación? Una empresa de productos sanitarios debe entender que nunca es demasiado pronto para empezar a trabajar en la validación, una empresa debe empezar a validar más pronto que tarde para averiguar que va por el camino correcto y que resuelve el problema correcto.
La validación (también V&V) al ser un proceso iterativo consume una buena inversión, cuando se planifica mal. Una estrategia de pruebas bien definida puede ayudarle a optimizar los costes y el periodo de pruebas para que el producto esté listo para el mercado a tiempo.
La complejidad de cualquier estrategia de pruebas depende de las tecnologías que se vayan a utilizar y de los mercados geográficos de destino. La estrategia de pruebas debe abarcar al menos los seis parámetros que se mencionan a continuación:
- Geografías objetivo y normas asociadas;
- Tiempo de comercialización;
- Una norma a seguir con una versión;
- Laboratorios de pruebas - laboratorios internos o independientes;
- Definición de la secuencia de pruebas;
- Presentación del resultado de la prueba
En consecuencia, las pruebas utilizadas para el proceso de verificación y validación también deben ser validadas. Esto es para asegurar que se mide lo que se necesita medir, porque una prueba equivocada dará resultados erróneos de usabilidad y funcionalidad. Las empresas de productos sanitarios necesitan un proceso de verificación y validación eficaz y bien documentado, que cumpla con la normativa asociada.
Diseño de dispositivos médicos: Gestión de riesgos
Estrategia de migración de riesgos vs. Plan de gestión de riesgos
Los procedimientos de gestión de riesgos de los productos sanitarios se rigen por una norma de cumplimiento internacionalmente aceptada ISO 14971:2007 Productos sanitarios - "Aplicación de la gestión de riesgos a los productos sanitarios". Aparte de esto, las políticas de gestión de riesgos deben incorporarse en todas las etapas de diseño y desarrollo de los productos sanitarios y deben asociarse también a los aspectos de control del diseño [10].
La gestión de riesgos nunca termina (¡al menos en teoría!). La filosofía de la gestión de riesgos es que no debe tratarse de un conjunto de reglas duras y rápidas. La gestión de riesgos y la estrategia de migración de riesgos consisten en comprender la intención de la gestión de riesgos y abordar el proceso de forma lógica y sistemática. En otras palabras, no te limites a seguir las normas... ¡piensa!
Considering the complexity of medical device design, focused risk management practices help ensure usability, safety, and cumplimiento de la normativa. It is a process of identifying, controlling, and preventing the failure that may cause hazards to users. It also mandates identifying associated risks.
La figura 3 muestra todas las etapas del proceso de gestión de riesgos. El proceso comienza con la identificación de los peligros y, a continuación, se mide el riesgo asociado en función de las consecuencias de los peligros y su posibilidad de riesgo.
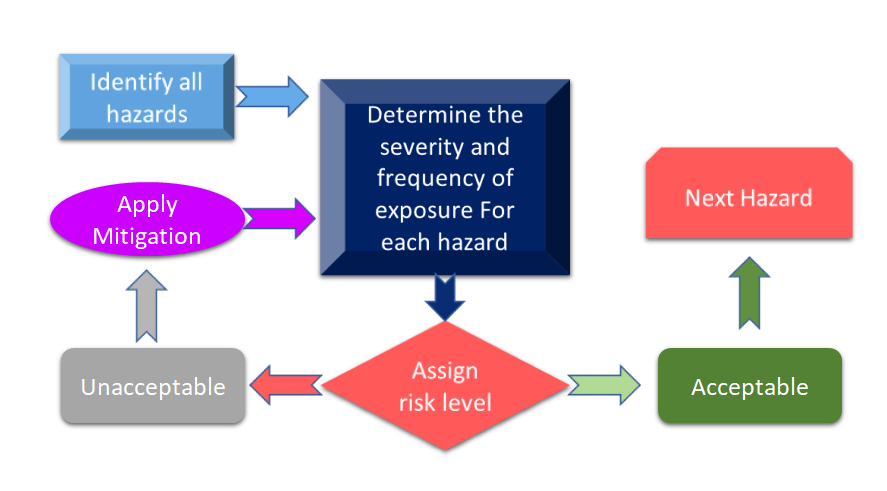
Figura 3: Proceso de gestión de riesgos para productos sanitarios (adaptado de [11]).
En caso de que el riesgo identificado durante el proceso de gestión de riesgos de los productos sanitarios supere los criterios definidos, será necesario aplicar una mitigación de riesgos. El nivel de riesgo depende de varios parámetros que incluyen, entre otros, su dispositivo, las tecnologías y, en algunos casos, la forma en que su empresa gestiona el proceso de mitigación de riesgos. Siempre es aconsejable realizar un análisis de riesgos para su dispositivo para ver qué normas pueden aplicarse a su dispositivo. En la reciente revisión de la norma ISO 14971 Norma Internacional para la Gestión de Riesgos de los Productos Sanitarios, el análisis de riesgos y el Análisis Preliminar de Peligros (PHA) se identifican como requisitos principales para su producto sanitario [12]. De manera simplificada, el PHA está destinado a proporcionar el marco inicial para las evaluaciones y la gestión de riesgos, y abarca tanto el análisis como la evaluación de riesgos. Según la definición, el PHA comprende una lista de peligros, daños y situaciones peligrosas, formulados a partir de los materiales de construcción (MdC) de sus dispositivos, los componentes o la materia prima utilizada en su dispositivo, y las interfaces hombre-dispositivo o manuales, el entorno de uso, el principio de funcionamiento y otros factores relevantes. [13].
Conclusión:
Al final, para cualquier empresa nueva de dispositivos médicos o para una organización bien establecida, es importante recordar que leer la normativa no le hace ganar nada, pero entender la filosofía le hace ganar mucho.
Conclusión: cuando se trata de analizar y planificar los riesgos:
- Debe utilizarse desde el principio y durante todo el proceso de diseño y desarrollo,
- A menudo se genera nueva información para retroalimentar el proceso de diseño y desarrollo (de dispositivos actuales/futuros),
- Ninguna planificación puede eliminar todos los peligros y riesgos... pero sí puede mitigar muchos de ellos. (Seguir las filosofías de control del diseño descritas aquí mitiga automáticamente los riesgos).
La ruta de acceso al mercado de cada producto sanitario es compleja debido a los diversos factores que hay que tener en cuenta, como los patrones de uso, el material, la experiencia del usuario y la normativa, entre otros.
Necesito ayuda con medical device design? Browse experienced expertos del sector de la tecnología médica en Kolabtree o publique su proyecto de forma gratuita para obtener propuestas.
REFERENCIAS Y RECURSOS
- https://www.welldoc.com/health-plans/
- https://ec.europa.eu/docsroom/documents/10337/attachments/1/translations
- FDA, 2005, Total Product Lifecycle, FDA-CDRH Presentation by CDRH Director Dr. David Feigal, http://www.fda.gov/cdrh/strategic/presentations/ tplc.html
- Pietzsch, Jan & Shluzas, Lauren & Paté-Cornell, Marie-Elisabeth & Yock, Paul & Linehan, John. (2009). Stage-Gate Process for the Development of Medical Devices. Journal of Medical Devices. 3(2).
- Estrategias normativas para la tercera edición de la norma IEC 60601-1 Recuperado el 9 de septiembre de 2020.
- https://www.meddeviceonline.com/doc/an-introduction-to-international-medical-device-standards-0001
- https://www.fda.gov/files/drugs/published/Design-Controls—Devices.pdf
- Feigal DW. Apéndice D. Impact of the Regulatory Framework on Medical Device Development and Innovation. Comité del Instituto de Medicina (EE.UU.) sobre el Salud pública Effectiveness of the FDA 510(k) Clearance Process; Wizemann T, editor. Public Health Effectiveness of the FDA 510(k) Clearance Process: Balancing Patient Safety and Innovation: Workshop Report. Washington (DC): National Academies Press (US); 2010. Appendix D, Impact of the Regulatory Framework on Medical Device Development and Innovation. Available from: https://www.ncbi.nlm.nih.gov/books/NBK209794/.
- 1997, FDA CDRH 1997, Design Control Guidance for Medical Device Manufacturers
- https://starfishmedical.com/blog/iso-134852016-section-7/?doing_wp_cron=1599995964.4528369903564453125000
- Teixeira, M. B., y Bradley, R., 2003, Design Controls for the Medical Device Industry, Marcel Dekker, Nueva York.
- ISO 14971:2019 - Medical devices - Application of risk management to medical devices
- ISO/TR 24971:2020 - Medical devices - Guidance on the application of ISO 14971
Todos los artículos de esta serie:
Desarrollo y diseño de dispositivos médicos: Una guía definitiva
Desarrollo de dispositivos médicos: 3 consejos para el éxito
Diseño de dispositivos médicos: La guía esencial, paso a paso
Comercialización de dispositivos médicos: 9 pasos desde el boceto hasta el lanzamiento
Cómo superar los retos de la comercialización de dispositivos médicos
Lanzamiento de dispositivos médicos: Pasos clave para llevar su producto al mercado
Vigilancia postcomercialización de dispositivos médicos: Una guía completa
Kolabtree helps businesses worldwide hire freelance scientists and industry experts on demand. Our freelancers have helped companies publish research papers, develop products, analyze data, and more. It only takes a minute to tell us what you need done and get quotes from experts for free.

Unlock Corporate Benefits
• Secure Payment Assistance
• Onboarding Support
• Dedicated Account Manager
Sign up with your professional email to avail special advances offered against purchase orders, seamless multi-channel payments, and extended support for agreements.


