



Nare Simonyan, freelance regulatory affairs specialist de Kolabtree, ofrece una guía completa sobre un expediente reglamentario y su formato.
Introducción: ¿Qué es un expediente normativo?
Regulatory dossier is a package of documents, which may include all required information regarding newly developed drug products and/or generics, which is required by EU and US regulatory authorities for granting marketing authorization approvals. The main information that is included in the package is administrative information, data related to the quality, safety and efficacy of drug product, which can be submitted by CTD (Common Technical Document) format both paper and electronic version. Since the information submitted in paper format was enormous, agencies are now encouraging applications to be submitted in eCTD format.
CTD (Documento Técnico Común)
En 2003, los miembros de la ICH (Consejo Internacional de Armonización) acordaron reunir toda la información sobre calidad, seguridad y eficacia en un formato común que se denominaba CTD. El CTD es un formato/estructura para los módulos 1 a 5 de la NDA (solicitud de nuevo fármaco), la MAA (solicitud de autorización de comercialización) y las solicitudes globales de medicamentos. El módulo 1 contiene información administrativa regional que es diferente para cada país. Los módulos 2, 3, 4 y 5 son comunes para todas las regiones. La estructura del CTD se aplica tanto a las solicitudes en investigación como a las comerciales (IND (Investigational New Drug) y NDA (US); IMPD (Investigational Medicinal Product Dossier) y MAA (EU), y solicitudes globales).
The CTD was primarily used for new marketing applications such as NDA, BLA (Biologics License Application), MAA, NDS (New Drug Substance), JNDA (Japanese New Drug Application), etc. With additional guidance and guidelines from the FDA and EMA, the CTD is now required for all applications, including those for ensayos clínicos—IMPD and INDs. All Drug Master Files (DMF) and Active Substance Master Files (ASMF) must follow the structure of the CTD. The process of submitting regulatory dossier is regulated by Code of Federal Regulations (CFR) (Law in US) and Directives (Law in EU).
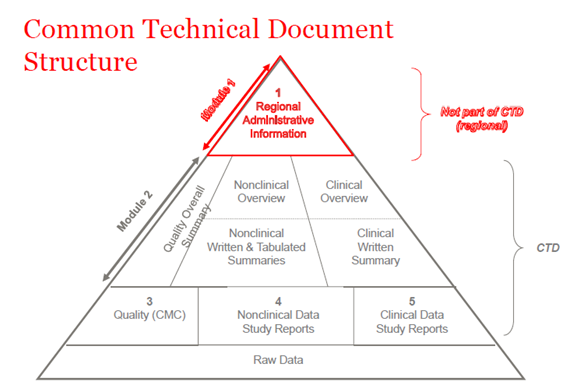
Figura 1. La estructura del CTD
Módulo CTD 1
Especificaciones para el Módulo 1 de EE.UU.: Información administrativa e información de prescripción
La sección del Módulo 1 del e-CTD contiene documentos administrativos y de etiquetado. Todas las solicitudes y presentaciones relacionadas tienen la misma estructura organizativa para los documentos del Módulo 1.
A continuación, encontrará algunas especificaciones sobre los documentos que debe proporcionar en el módulo 1:
Carta de presentación (sección 1.2)
Las cartas de presentación contienen información pertinente que ayuda a la comunicación en el proceso de revisión. Se recomienda incluir la siguiente información en la carta de presentación (consulte "Documento Técnico Común Electrónico (eCTD) v4.0 GUÍA DE CONFORMIDAD TÉCNICA")
- Descripción reglamentaria de la presentación, incluyendo la información reglamentaria apropiada, y cualquier hipervínculo deseado a la información presentada
- Descripción técnica del envío, incluyendo el tamaño aproximado del mismo (por ejemplo, 2 gigabytes)
- Declaración de que el envío está libre de virus, con una descripción del software (nombre, versión y empresa) que se utilizó para comprobar los archivos en busca de virus
- Un punto de contacto reglamentario y técnico para la presentación, incluida la dirección de correo electrónico.
Certificación de copia de campo (sección 1.3)
La certificación de la copia de campo debe incluirse en el eCTD para las solicitudes de comercialización. Puede ser una carta dirigida a la oficina de distrito en la que se notifique que se presentará el eCTD a la FDA. La carta debe incluir:
- Número de medicamento y de solicitud
- Centro y división de la FDA
- La solicitud está en formato eCTD.
Referencias (sección 1.4)
Las referencias pueden contener los siguientes subapartados (también se pueden exigir contenidos adicionales)
- Cartas de autorización (LOA)
- Referencias cruzadas de información presentada anteriormente que no está en formato eCTD
Modificaciones de la información (sección 1.11)
Puede utilizarse para la presentación de respuestas a solicitudes de información (IR), cuando la información que se presenta no encaja en ningún epígrafe de los módulos 2, 3, 4 o 5. Si la respuesta a la solicitud de información afecta a los documentos presentados en los módulos 2 a 5, los documentos nuevos o sustitutivos deberán presentarse en el lugar correspondiente del módulo 2 a 5 y se hará referencia a ellos en el documento de la sección 1.11.
Informes anuales de marketing (sección 1.13)
Para cada estudio o ensayo descrito en los expedientes de requisitos/compromisos de postcomercialización debe incluirse un marcador.
Etiquetado (sección 1.14)
La sección 1.14 describe cómo proporcionar documentos de etiquetado específicos. Puede incluir el borrador del etiquetado, el etiquetado final, el etiquetado de medicamentos listados, el etiquetado de medicamentos en investigación, el etiquetado extranjero y el etiquetado del producto para las presentaciones 2253*. La información proporcionada puede incluir la historia, el contenido y las muestras del etiquetado.
Formulario FDA 2253*: Transmisión de la publicidad y el etiquetado promocional de medicamentos y productos biológicos de uso humano
Publicidad y material de etiquetado promocional (Sección 1.15)
La publicidad y el etiquetado promocional están restringidos en EE.UU., y deben reflejar los requisitos mencionados en la Guía de la FDA Suministro de presentaciones reglamentarias en formato electrónico y no electrónico - Etiquetado promocional y material publicitario para medicamentos de prescripción humana.
En el caso de los materiales promocionales presentados como parte de los requisitos de información posterior a la comercialización, se sugiere que se proporcionen enlaces de hipertexto a las referencias o al etiquetado. Las referencias mejoran la eficacia de una revisión.
Estrategia de Evaluación y Mitigación de Riesgos (REMS) (Sección 1.16)
El suplemento del REMS está destinado a proponer un nuevo REMS o modificaciones (mayores y/o menores) a un REMS aprobado. En el formulario de presentación de la FDA apropiado debe seleccionarse el tipo de suplemento de REMS. Las evaluaciones de REMS, las revisiones de REMS y las correspondencias de REMS no son suplementos. En este caso tipo de presentación debe seleccionarse "otros" al rellenar el formulario de la FDA.
Especificaciones para el Módulo 1 de la Unión Europea (UE): Información administrativa e información de prescripción
Carta de presentación (sección 1.0)
Para cada solicitud debe presentarse una carta de presentación. Los documentos "Notas a los revisores" pueden incluirse como apéndice de la carta de presentación, en caso de que sea necesario proporcionar más información para facilitar la navegación.
Índice completo (sección 1.1)
Para cada tipo de solicitud se debe presentar un índice completo, que puede contener todas las secciones del módulo que se han presentado como parte de la solicitud en cuestión. En el caso de las nuevas solicitudes, deberán abordarse todas las secciones.
Formulario de solicitud (sección 1.2)
Dependiendo del tipo de presentación, el formulario de solicitud correspondiente debe incluirse en el expediente reglamentario.
Los diferentes formularios de solicitud están disponibles en el sitio web de la Comisión Europea /
DG Empresa:
- Nuevas solicitudes y solicitudes de ampliación http://ec.europa.eu/enterprise/pharmaceuticals/eudralex/homev2.htm#2b
- Solicitudes de variación
http://ec.europa.eu/enterprise/pharmaceuticals/eudralex/homev2.htm#2c
- Solicitudes de renovación
http://ec.europa.eu/enterprise/pharmaceuticals/eudralex/homev2.htm#2c
Información sobre el producto (sección 1.3)
La sección 1.3 contiene información sobre
- Resumen de las características del producto (RCP), etiquetado y prospecto (PIL) (Sección 1.3.1)
- Maqueta (Sección 1.3.2)
- Muestra (Sección 1.3.3)
- Consulta con los grupos de pacientes destinatarios (Sección 1.3.4)
- Información sobre el producto ya aprobado en los Estados miembros (Sección 1.3.5) (si procede)
- Braille (Sección 1.3.6)
Información sobre los expertos (sección 1.4)
En esta sección es necesario incluir la información relativa a los expertos con informes detallados de los documentos e información que constituyen los módulos 3, 4 y 5 de acuerdo con el artículo 12 de la Directiva 2001/83 / CE. Para la parte adicional de esta sección se puede utilizar un informe de expertos firmado para las diferentes partes científicas del expediente. Los requisitos de los informes de expertos firmados se presentan a continuación:
- El resumen general de calidad, el resumen no clínico y el resumen clínico del módulo 2,
- Una declaración firmada por los expertos del módulo 1.4.
- Una breve información sobre el historial educativo, la formación y la experiencia profesional en el módulo 1.4.
En el caso de las solicitudes posteriores a la autorización, se debe presentar la declaración o declaraciones de los expertos correspondientes.
En los casos en que los titulares de la autorización de comercialización deseen distinguir dicha declaración de cualquier
declaraciones anteriores, el número de procedimiento correspondiente del Estado miembro/EME de referencia
puede incluirse en la parte superior.
A continuación se presentan subsecciones de información sobre los expertos:
- Calidad (sección 1.4.1)
- No clínico (1.4.2)
- Clínica (Sección1.4.3)
Para más información y las plantillas correspondientes, consulte el artículo 12 y de acuerdo con el anexo I, parte 1.4 de la Directiva 2001/83/CE.
Requisitos específicos para los distintos tipos de aplicaciones (sección 1.5)
Información para aplicaciones bibliográficas (Sección 1.5.1)
Basándose en el artículo 10 bis de la Directiva 2001/83/CE, los solicitantes deben presentar un documento conciso que resuma los motivos y las pruebas utilizadas para demostrar que los componentes del medicamento tienen un uso bien establecido, teniendo en cuenta el nivel aceptable de seguridad y eficacia, tal como se indica en la parte II.1 del anexo I de la Directiva 2001/83/CE.
Información para solicitudes de genéricos, "híbridos" o biosimilares (sección 1.5.2)
Basándose en los apartados 1, 3 o 4 del artículo 10 de la Directiva 2001/83/CE, los solicitantes deben presentar aquí un documento conciso que resuma los motivos y las pruebas utilizadas para demostrar el tipo de medicamento para el que se presenta la solicitud. Los medicamentos pueden ser genéricos, productos híbridos y biosimilares.
(Ampliado) Datos / Exclusividad de mercado (Sección 1.5.3)
La sección 1.5.3 es necesaria cuando el titular/solicitante de la autorización de comercialización desea reclamar datos/exclusividad comercial (adicionales) mientras solicita una nueva indicación o un cambio de clasificación. En este caso, deben tenerse en cuenta las disposiciones y los requisitos legales pertinentes.
Circunstancias excepcionales (sección 1.5.4)
Según el artículo 22 de la Directiva 2001/83/CE y el apartado 7 del artículo 14 del Reglamento (CE) nº 726/2004, se puede conceder una autorización en caso de circunstancias excepcionales, cuando el solicitante introduzca procedimientos específicos, en particular en lo que respecta a la seguridad del medicamento y a la notificación a las autoridades competentes de cualquier incidente relacionado con su uso. Sólo en el caso de excepciones objetivas y verificables puede concederse una autorización.
Autorización de comercialización condicionada (sección 1.5.5)
Esta sección es aplicable al procedimiento centralizado. Las referencias para esta sección son el apartado 7 del artículo 14 del Reglamento (CE) nº 726/2004 y "Directrices sobre la aplicación científica y las modalidades prácticas de la autorización condicional de comercialización".
Evaluación del riesgo medioambiental (sección 1.6)
De conformidad con las letras c bis) y g) del artículo 8 de la Directiva 2001/83/CE, el solicitante debe tener en cuenta cualquier riesgo potencial del medicamento para el medio ambiente al solicitar la aprobación de la autorización de comercialización. Los requisitos de la Directiva están relacionados con el uso, el almacenamiento y la eliminación de los medicamentos, y no son aplicables a la síntesis o la fabricación del producto. Las solicitudes de autorización de comercialización de medicamentos que no contengan OGM (Sección 1.6.1 de no OGM) y que sí contengan OGM (1.6.2 de OGM) deben incluir una indicación de cualquier riesgo potencial en el módulo 1 del expediente reglamentario.
Para más información, consulte "Directriz sobre la evaluación del riesgo medioambiental de los medicamentos de uso humano"
Información relativa a la exclusividad del mercado huérfano (sección 1.7)
Esta sección sólo es aplicable a los medicamentos huérfanos. La información necesaria sobre los detalles y el procedimiento están presentes en "Directriz de la Comisión Europea sobre aspectos de la aplicación del artículo 8 del Reglamento (CE) nº 141/2000: Evaluación de la similitud y/o superioridad clínica de los medicamentos huérfanos al evaluar las solicitudes de autorización de comercialización y las variaciones."
Son aplicables la sección 1.7.1 de similitud y la sección 1.7.2 de exclusividad comercial, si el medicamento huérfano ha sido autorizado para la condición que cubre la indicación terapéutica propuesta que se solicita, y está en vigor un periodo de exclusividad comercial, los solicitantes deben presentar un informe crítico que aborde la posible similitud con el medicamento huérfano autorizado y que concluya sobre la similitud o "no" similitud (1.7.1). Si el medicamento objeto de la solicitud de autorización de comercialización se considera "similar" a un medicamento huérfano cubierto por las disposiciones de exclusividad comercial antes mencionadas, el solicitante deberá además justificar que una de las excepciones establecidas como se describe en el artículo 8.3, párrafos (a) a (c) del Reglamento (CE) nº 141/2000(1.7.2).
Information relating to Farmacovigilancia (Sección 1.8)
Sistema de farmacovigilancia (sección 1.8.1)
La farmacovigilancia es la ciencia y las actividades relacionadas con la detección, evaluación, comprensión y prevención de los efectos adversos o cualquier otro problema relacionado con los medicamentos.
Esto se aplica durante todo el ciclo de vida del medicamento, tanto en la fase previa a la aprobación como en la posterior. El sistema de farmacovigilancia es un apartado muy importante para solicitar la autorización de comercialización.
Debe proporcionarse una descripción detallada del sistema de farmacovigilancia de acuerdo con el artículo 8 (ia) de la Directiva 2001/83/CE. La descripción debe incluir la prueba de que el solicitante cuenta con los servicios de una persona cualificada responsable de la farmacovigilancia.
La descripción del sistema de farmacovigilancia del titular de la autorización de comercialización debe seguir los requisitos y el formato detallados en el volumen 9A de EudraLex.
Sistema de gestión de riesgos (sección 1.8.2)
Detailed description of risk-management system must be provided according to Article 8 (ia) of Directive 2001/83/EC. The detailed description of a risk management system should be provided in the form of an EU Risk Management Plan (EU-RMP), as outlined in Volume 9A of EudraLex.
Información relacionada con los ensayos clínicos (sección 1.9)
La sección de 1.9 debe prepararse de acuerdo con el artículo 8 (ib) de la Directiva 2001/83/CE para informar de que los ensayos clínicos realizados fuera de la Unión Europea cumplen los requisitos éticos de la Directiva 2001/20/CE, si procede.
Esta sección debe proporcionarse para todas las nuevas solicitudes (incluidas las solicitudes de extensión), y otros procedimientos regulatorios relevantes posteriores a la autorización (por ejemplo, variaciones) para los cuales se presentaron informes de ensayos clínicos.
Información relativa a la pediatría (sección 1.10)
De conformidad con los artículos 7, 8 y 30 del Reglamento (CE) nº 1901/2006 ("Reglamento pediátrico") y el artículo 23 del Reglamento (CE) nº 1901/2006 ("Reglamento pediátrico"), esta sección es obligatoria:
- Para todas las nuevas solicitudes*, para un medicamento que no esté autorizado en el EEE
- Para las solicitudes* de nuevas indicaciones, nuevas formas farmacéuticas y nuevas vías de administración, para los medicamentos autorizados que estén protegidos por un certificado de protección complementaria, o por una patente que permita la concesión de dicho certificado.
- Para las solicitudes de autorización de comercialización de uso pediátrico (PUMA)
*excepto en el caso de las aplicaciones genéricas, híbridas, biosimilares y de uso bien establecido y de los medicamentos tradicionales a base de plantas u homeopáticos
Para el módulo 1 se puede proporcionar:
Respuestas a las preguntas en los casos en que se aconseja a los solicitantes que incluyan en esta sección un documento que enumere las preguntas con la correspondiente respuesta de texto narrativo para cada pregunta, y cuando las respuestas también contengan datos/documentos nuevos o actualizados relacionados con los Módulos 3, 4 y/o 5. Dichos datos/documentos deberán colocarse en las secciones correspondientes de dichos Módulos.
Datos adicionales. Esta sección es necesaria según el procedimiento de autorización. Es posible que sea necesario proporcionar datos adicionales como parte de una solicitud nacional, descentralizada o de reconocimiento mutuo. Si dichos datos están relacionados con los Módulos 2, 3, 4 y/o 5, los documentos también deben colocarse en las secciones pertinentes de dichos Módulos. Los requisitos específicos de los Estados miembros en materia de datos adicionales pueden consultarse en el sitio web de la Comisión Europea.
Módulo CTD 2
La introducción general al medicamento se presenta en la sección del Módulo 2 del expediente CTD, que está armonizado para todas las regiones (Consejo Internacional para la Armonización de los Requisitos Técnicos para Productos Farmacéuticos de Uso Humano (ICH)). Este módulo se presenta mediante documentos de resumen para cada uno de los módulos siguientes: datos de calidad, informes de estudios no clínicos y clínicos.
Módulo CTD 3
Module 3 was identified by ICH as the Quality Module. Thus, “Quality” became the global term for CMC (química, manufacturing and controls). La CMC no debe confundirse con los elementos de control de calidad, la garantía de calidad, los PNT (procedimientos operativos estándar), los documentos internos de la empresa (especificaciones, registros de lotes, etc.)
La sección del módulo 3 también está armonizada para todas las regiones con el suministro de información químico-farmacéutica y biológica para las sustancias químicas activas y los medicamentos biológicos.
A lo largo del desarrollo y después de que un producto sea aprobado por las autoridades sanitarias, los aspectos de química, fabricación y control (CMC) siguen evolucionando y cambiando.
Calidad farmacéutica = CMC
QUÍMICA
Descubrimiento de nuevas entidades químicas/moléculas, purificación de principios activos (sustancia farmacéutica), pruebas analíticas de materias primas y productos acabados.
FABRICACIÓN
Fabricación a escala de laboratorio de sustancias y productos farmacéuticos, fabricación de suministros clínicos para estudios clínicos, ampliación a tamaño de lote comercial, producto comercial.
Y CONTROLES
Desarrollar especificaciones/controles adecuados para la sustancia y el producto farmacéutico a fin de garantizar la seguridad, la eficacia y la calidad.
Todas las regiones exigen un expediente de CMC/Calidad Farmacéutica
- Iniciar y realizar ensayos clínicos,
- Para presentar un nuevo expediente de autorización de comercialización (MAA, NDA, BLA, etc.)
- Cumplir los requisitos reglamentarios para la gestión del ciclo de vida y los cambios posteriores a la aprobación del producto
Este módulo incluye todo el paquete de documentos relativos a la sustancia farmacéutica (DS) y al producto farmacéutico (DP).
Sustancia de la droga (DS)
La sección de sustancias farmacéuticas se presenta mediante su descripción, incluyendo las características físicas, químicas o biológicas, el nombre y la dirección del fabricante de DS, los límites aceptables y los métodos analíticos utilizados para garantizar la identidad, la potencia, la calidad y la pureza de la sustancia farmacéutica.
También se incluye información que respalde la estabilidad del fármaco durante los estudios toxicológicos y el estudio clínico propuesto.
PRODUCTO FARMACÉUTICO (DP)
En esta sección se describe una lista de todos los componentes, que puede incluir alternativas razonables para los compuestos inactivos, utilizados en la fabricación del producto farmacéutico, incluyendo tanto los componentes que están destinados a aparecer en el producto farmacéutico como los que pueden no aparecer, pero que se utilizan en el proceso de fabricación.
A continuación, se enumeran los datos clave del producto farmacéutico que deben incluirse en el expediente reglamentario:
- La composición cuantitativa del medicamento (en investigación o comercial), incluyendo cualquier variación razonable.
- El nombre y la dirección del fabricante del medicamento.
- Descripción de los procedimientos de fabricación y envasado.
- Los límites aceptables y los métodos analíticos utilizados para asegurar la identidad, la potencia, la calidad y la pureza del producto farmacéutico.
Módulo CTD 4
Informes de estudios no clínicos
La sección pertinente la ubicación adecuada para los datos de los animales individuales es en el informe del estudio en el Documento Técnico Común para las solicitudes que se presentarán a las Autoridades Reguladoras. El Módulo 4 está armonizado para EE.UU. y la UE sobre la base de los principios de la ICH, y contiene todas las secciones y subsecciones necesarias para los informes de los estudios. Referencias a las directrices de la ICH Documento Técnico Común (CTD).
Módulo CTD 5
Informes de estudios clínicos
La sección del módulo 5 es la estructura y el contenido de los informes de estudios clínicos. Esta parte del CTD presenta los informes de estudios humanos/clínicos, otros datos clínicos y referencias dentro de un Documento Técnico Común (CTD) para el registro de un producto farmacéutico de uso humano. Estos elementos deben facilitar la preparación y revisión de una solicitud de comercialización. El módulo 5 está armonizado para EE.UU. y la UE sobre la base de los principios de la ICH, y contiene todas las secciones y subsecciones necesarias para los informes de estudios. Referencias a las directrices de la UE Documento Técnico Común (CTD).
¿Necesita ayuda para preparar un expediente normativo? Ver y consultar redactores autónomos de normativa en Kolabtree.
Referencias adicionales
- https://www.ich.org/page/ctd
- https://www.fda.gov/media/128163/download
- https://www.fda.gov/media/135573/download
- https://ec.europa.eu/health/sites/default/files/files/eudralex/vol-2/b/update_200805/ctd_05-2008_en.pdf
- https://ec.europa.eu/health/sites/default/files/files/eudralex/vol-1/dir_2001_83_consol_2012/dir_2001_83_cons_2012_en.pdf
- https://eur-lex.europa.eu/legal-content/EN/TXT/PDF/?uri=CELEX:02001L0083-20121116&from=DE
- https://eur-lex.europa.eu/legal-content/EN/TXT/PDF/?uri=CELEX:02004R0726-20130605&from=EN
- https://ec.europa.eu/health/sites/default/files/files/eudralex/vol-1/reg_2000_141_cons-2009-07/reg_2000_141_cons-2009-07_en.pdf
- https://www.ema.europa.eu/en/human-regulatory/research-development/clinical-trials-human-medicines
- https://eur-lex.europa.eu/legal-content/EN/TXT/?uri=celex%3A32006R1901
- https://clinicaltrials.gov/ct2/home
Kolabtree helps businesses worldwide hire freelance scientists and industry experts on demand. Our freelancers have helped companies publish research papers, develop products, analyze data, and more. It only takes a minute to tell us what you need done and get quotes from experts for free.

Unlock Corporate Benefits
• Secure Payment Assistance
• Onboarding Support
• Dedicated Account Manager
Sign up with your professional email to avail special advances offered against purchase orders, seamless multi-channel payments, and extended support for agreements.


