



Shreya Chenni, freelance regulatory writer for medical devices, provides a 10-minute guide to FDA design controls for your medical device.
One of the major causes of medical device recalls is the lack of design control as identified by the FDA [3,3a]. The pre-production controls were then added to the device GMP regulations. Design controls are an interrelated set of practices and procedures that are incorporated into the design and development process, i.e., a system of checks and balances. Design controls increase the likelihood that the design transferred to production will translate into a device that is appropriate for its intended use.
Applicability
All Class II, III and the following Class I devices are subjected to design controls:
- Devices automated with computer software
- 868.6810 Catheter, Tracheobronchial Suction
- 878.4460 Glove, Surgeon’s
- 880.6760 Restraint, Protective
- 892.5650 System, Applicator, Radionuclide, Manual
- 892.5740 Source, Radionuclide Teletherapy
- Devices automated with computer software
- Tracheobronchial suction catheters
- Surgeon’s gloves
- Protective restraints
- System, radionuclide, applicator, manual
- Source, radionuclide teletherapy
Design controls apply to all D&D activities – for novel or improved devices being developed in the pre-market phase, as well as for changes to existing, marketed devices. Design controls do not apply to research activities conducted during the proof of concept stage.
Design controls can be applied to any product development process. The following flowcharts are examples of the design controls applied to a traditional Waterfall design process and V-model process for Software (SW).
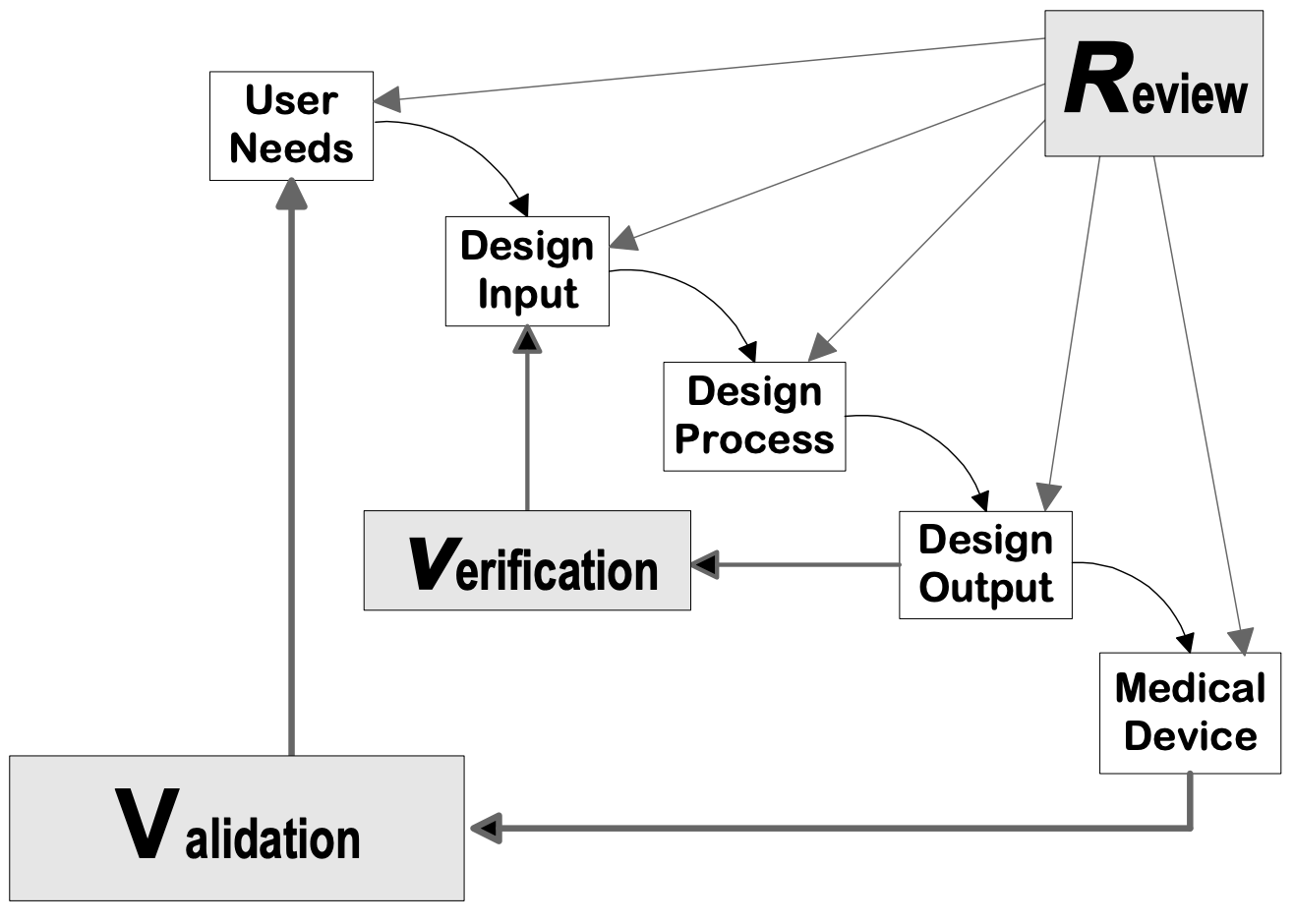
Source: Design Control Guidance for Medical Device Manufacturers, CDRH-FDA
Device Design Phases
D&D Planning Phase
D&D plans should be established and maintained. The plan shall describe or reference the design and development activities and assign responsibilities for implementation. At a minimum the FDA recommends on including the following in the plan:
- Goals and objectives of the D&D program
- Delineation of responsibilities of the design activities
- Identify major tasks, deliverables and assign responsibility for each task
- Scheduling of major task in accordance with the main development timeframe
- Identify the major reviews and decision points
- Identify reviewers, reviewing team and the procedures to be followed by reviewers
- Controls of design documentation
- Notification activities
The plan should be reviewed, updated, and approved as design and development evolves.
Design Input Phase
This is the starting point for product design. The medical device is designed and developed to meet the user requirements. Gather the user requirements from various sources such as customer surveys, feedback from the physicians, complaints. These requirements are transferred into design inputs.
- Design inputs: These are the physical and performance requirements of a device that are used for device design. The design input phase consists of the converting user requirements into product requirements. Regulatory requirements are considered when defining design inputs. Design input requirements must be comprehensive, unambiguous and verifiable objectively. The inputs requirements can be put together into 3 categories:
- Functional requirements, that describe what the device does. For example: The wheelchair shall move forward when indicated by the user
- Performance requirements, which will specify how much and how well the device should perform. For example: The wheelchair shall move with a speed of 2m/s in the forward direction
- Interface requirements, specify characteristics of the device which are critical to compatibility with external systems, such as user/patient interface. For example: The wheelchair shall have buttons with direction indicating symbols
- Here is an example, which defines user needs and converts it to design input:
- User need
Device should be portable and bluetooth enabled
- User need
-
- Design input
Identify the applicable FDA recognised or an international standard to be compliant with. Such as:
-
-
- IEEE ANSI C63.27-2017 American National Standard for Evaluation of Wireless Coexistence
- AAMI TIR 69: Association for the Advancement of Medical Instrumentation – Risk Management of Radio-frequency Wireless Coexistence for Medical Devices and Systems (2017)
- IEC 60601-1-2 Edition 3: 2007: Medical Electrical Equipment – Part 1-2: General Requirements for Safety – Collateral Standard: Electromagnetic Compatibility – Requirements and Tests
- UL 2054 – Standard for Household and Commercial Batteries
-
List the performance and other specific inputs. Such as:
-
-
- The device shall be DC powered
- Bluetooth module shall be used
- Weight: approx 6lbs or 6lbs+/- 2lbs (Inputs should have quantitative limits to ensure verification)
-
Design Output Phase
Design outputs are the results of a design effort at each design phase and at the end of the total design effort. Examples of design outputs are engineering drawings, labeling, work instructions and other product specifications. Other design outputs include results of risk analysis, results of verification activities, biocompatibility test results and software source code.
Design outputs should not be released before review and approval by the responsible personnel. It should also be noted that any changes to the device after the approval of design outputs/inputs will be controlled through review and approval by concerned personnel. A design review is required at the end of this phase.
Design Review Phase
A design review should be held after the design output phase. The design reviews should follow established procedures and be documented in the Design History File (DHF). The participants of each design review should have representatives from all functional groups.
Formal reviews are recommended to be conducted at the end of important project milestones. Commonly, design reviews are conducted after the design output phase, V&V phase and design transfer phanse. This also depends on the complexity of the device development. FDA requires at least one design review.
Design Verification Phase
Design verification is confirmation by objective evidence that design output meets design input. Basically, it is Design Input = Design Output. The verification activities should be performed per the established procedures. Examples of verification include EMC and electrical testing, visual inspection, non-clinical testing activities, fault tree analysis of process or design and failure modes and effects analysis. Verification ensures that the product specification technical requirements are met. All the verification activities have to be documented.
Traceability Matrix: This document consists of design inputs and outputs listed in a tabular format. For each input the corresponding output is referenced. This method of verification is used when inputs and outputs are both documents.
Design Validation Phase
Design Validation means establishing by objective evidence that specifications (specified requirements) conform with user needs and intended use(s).
- Process Validation means establishing by objective evidence that a process consistently produces a result or product meeting its predetermined specifications.
- Design Validation means establishing by objective evidence that device specifications conform with user needs and intended use(s).
Validation is typically conducted under actual or simulated conditions. Examples of validation include clinical trials, clinical evaluation, human factors tests, address packaging and labeling, analysis and inspections. Results of validation activities and/or validation reports should be documented which will be part of the Design History File (DHF).
Design Transfer Phase
After the completion of V&V stage, the design transfer takes place. This includes the transfer of device design into product specifications ensuring device quality. This phase is very critical because once the production of the device begins it will be subjected to design change control and can result in financial loss if problems are encountered. Design transfer should take place per the established procedure. Also, it should be ensured that documents that include product specifications are reviewed and approved before initiating design transfer.
Design Changes Phase
Design change control begins with design transfer and continues throughout the lifecycle. Any design change after design transfer will result in an Engineering Change Notice (ECN) to be carried out per an established procedure. It should be ensured that with any design change, related documents such as risk management report, instructions for use, verification and validation reports must be re-visited and updated as well.
Design History File (DHF)
The DHF is specific to the US FDA. ISO 13486:2016 doesn’t require the manufacturer to maintain a DHF.
A Design History File is maintained for each project which includes all the deliverables of each phase. It includes the latest product information. The design and development documents shall be easily available and accessed as and when needed. Design and development contracts should explicitly specify the manufacturer’s right to design information and establish standards for the form and content of design documentation.
In practice, design controls provide managers and designers with improved visibility of the design process. With improved visibility, managers are empowered to more effectively direct the design process—that is, to recognize problems earlier, make corrections, and adjust resource allocations. Designers benefit both by enhanced understanding of the degree of conformance of a design to user and patient needs, and by improved communications and coordination among all participants in the process.
Need help understanding and implementing FDA Design Controls for your medical device? Contact experienced medical device consultants on Kolabtree.
References:
- FDA, Design Control guidance for Medical Device Manufacturers
- Medical Regulatory Practices, An international perspective, Val Theisz
- Design Control, presentation by Joseph Tartal
3a. Federal Register / Vol. 61, No. 195
Kolabtree helps businesses worldwide hire freelance scientists and industry experts on demand. Our freelancers have helped companies publish research papers, develop products, analyze data, and more. It only takes a minute to tell us what you need done and get quotes from experts for free.

Unlock Corporate Benefits
• Secure Payment Assistance
• Onboarding Support
• Dedicated Account Manager
Sign up with your professional email to avail special advances offered against purchase orders, seamless multi-channel payments, and extended support for agreements.


