



Nare Simonyan, freelance regulatory affairs specialist na Kolabtree, fornece um guia abrangente para um dossiê regulamentar e seu formato.
Introdução: O que é um dossiê regulamentar?
Regulatory dossier is a package of documents, which may include all required information regarding newly developed drug products and/or generics, which is required by EU and US regulatory authorities for granting marketing authorization approvals. The main information that is included in the package is administrative information, data related to the quality, safety and efficacy of drug product, which can be submitted by CTD (Common Technical Document) format both paper and electronic version. Since the information submitted in paper format was enormous, agencies are now encouraging applications to be submitted in eCTD format.
CTD (Documento Técnico Comum)
Em 2003, os membros do ICH (Conselho Internacional de Harmonização) concordaram em reunir todas as informações de qualidade, segurança e eficácia em um formato comum que costumava ser chamado de CTD. O CTD é um formato/estrutura para os Módulos 1 a 5 do NDA (New Drug Application), MAA (Marketing Authorization Application), e aplicações médicas globais. O Módulo 1 contém informações administrativas regionais que são diferentes para cada país. Os Módulos 2, 3, 4 e 5 são comuns para todas as regiões. A estrutura CTD se aplica tanto para aplicações investigativas quanto comerciais (IND (Investigational New Drug) e NDA (US); IMPD (Investigational Medicinal Product Dossier) e MAA (EU), e aplicações globais).
The CTD was primarily used for new marketing applications such as NDA, BLA (Biologics License Application), MAA, NDS (New Drug Substance), JNDA (Japanese New Drug Application), etc. With additional guidance and guidelines from the FDA and EMA, the CTD is now required for all applications, including those for ensaios clínicos—IMPD and INDs. All Drug Master Files (DMF) and Active Substance Master Files (ASMF) must follow the structure of the CTD. The process of submitting regulatory dossier is regulated by Code of Federal Regulations (CFR) (Law in US) and Directives (Law in EU).
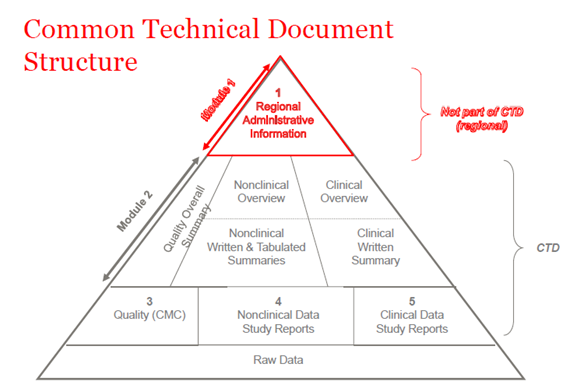
Figura 1. A estrutura do CTD
CTD-Módulo 1
Especificações para o Módulo 1 dos EUA: Informações Administrativas e Informações de Prescrição
A seção e-CTD Módulo 1 contém documentos administrativos e de etiquetagem. Todas as solicitações e apresentações relacionadas têm a mesma estrutura organizacional para documentos do Módulo 1.
Favor ver abaixo algumas especificações sobre documentos a serem fornecidos dentro do Módulo 1:
Carta de Apresentação (Seção 1.2)
As cartas de apresentação contêm informações pertinentes que ajudam a comunicação dentro do processo de revisão. As seguintes informações são recomendadas para serem incluídas na carta de apresentação (consulte "Documento Técnico Comum Eletrônico (eCTD) v4.0 GUIA DE CONFORMANÇA TÉCNICA”)
- Descrição regulamentar da apresentação, incluindo informações regulamentares apropriadas, e quaisquer hiperlinks desejados para as informações apresentadas
- Descrição técnica da apresentação, incluindo o tamanho aproximado da apresentação (por exemplo, 2 gigabytes)
- Declaração de que o envio é livre de vírus, com uma descrição do software (nome, versão e empresa) que foi utilizado para verificar a existência de vírus nos arquivos
- Um ponto de contato regulamentar e técnico para o envio, incluindo endereço de e-mail.
Certificação de cópia de campo (Seção 1.3)
A certificação de cópia de campo deve ser incluída no eCTD para aplicações de marketing. Pode ser uma carta ao escritório distrital notificando que a submissão do eCTD será submetida ao FDA. A carta deve incluir:
- Droga e número de aplicação
- Centro e divisão FDA
- A aplicação está no formato eCTD.
Referências (Seção 1.4)
As referências podem conter as seguintes subseções (conteúdo adicional também pode ser necessário)
- Cartas de Autorização (LOAs)
- Referenciamento cruzado Informações previamente enviadas que não estão no formato eCTD
Emendas de informação (Seção 1.11)
Isto pode ser usado para apresentação de respostas a Solicitações de Informação (RI), onde as informações que estão sendo apresentadas não se encaixam em nenhum título do Módulo 2, 3, 4 ou 5. Se a resposta do RI impactar documentos submetidos nos Módulos 2 - 5, os documentos novos ou substitutos devem ser submetidos ao local apropriado no Módulo 2 - 5 e referenciados no documento da Seção 1.11.
Relatórios anuais de marketing (Seção 1.13)
Para cada estudo ou ensaio descrito nos arquivos de requisitos/compromissos pós-comercialização, deve ser incluído um marcador de página.
Etiquetagem (Seção 1.14)
A seção 1.14 descreve como fornecer documentos de etiquetagem específicos. Pode incluir Rascunho de rotulagem, Rotulagem final, Rotulagem de drogas listadas, Rotulagem de drogas de investigação, Rotulagem estrangeira e Rotulagem de produtos para 2253 submissões*. As informações fornecidas podem conter o histórico, conteúdo e amostras da rotulagem.
Formulário FDA 2253*: Transmissão de anúncios e rotulagem promocional de drogas e produtos biológicos para uso humano
Anúncios e Material Promocional de Rotulagem (Seção 1.15)
Anúncios e materiais promocionais de rotulagem são restritos nos EUA, devem ser refletidos às exigências mencionadas na Orientação do FDA Fornecimento de Submissões Regulatórias em formato eletrônico e não-eletrônico - Materiais promocionais de rotulagem e publicidade para medicamentos de prescrição humana.
Para materiais promocionais enviados como parte das exigências de relatórios pós-comercialização, sugerimos que sejam fornecidos links de hipertexto para referências ou rótulos. As referências melhoram a eficiência de uma revisão.
Avaliação de Risco e Estratégia de Mitigação (REMS) (Seção 1.16)
O suplemento de REMS destina-se a propor um novo REMS ou modificações (maiores e/ou menores) a um REMS aprovado. No formulário apropriado do FDA, o suplemento REMS deve ser selecionado. Avaliações REMS, revisões REMS, e correspondências REMS não são suplementos. Neste caso, as tipo de apresentação deve ser selecionado "outro" ao preencher o formulário da FDA.
Especificações para a União Européia (UE) Módulo 1: Informações Administrativas e Informações de Prescrição
Carta de Apresentação (Seção 1.0)
Para cada solicitação, deve ser fornecida uma carta de apresentação. Os documentos "Notes to Reviewers" podem ser incluídos como Anexo à carta de apresentação, caso seja necessário fornecer mais informações para facilitar a navegação.
Índice abrangente (Seção 1.1)
Um índice completo deve ser fornecido para cada tipo de aplicação, que pode conter todas as seções de módulos que tenham sido apresentadas como parte da aplicação em questão. No caso de novos pedidos, todas as seções devem ser abordadas.
Formulário de Solicitação (Seção 1.2)
Dependendo do tipo de formulário de apresentação do pedido relevante deve ser incluído no dossiê regulamentar.
Os diferentes formulários de solicitação estão disponíveis no site da Comissão Européia /
DG Empresa:
- Novas Aplicações e Aplicações de Extensão http://ec.europa.eu/enterprise/pharmaceuticals/eudralex/homev2.htm#2b
- Aplicações de variação
http://ec.europa.eu/enterprise/pharmaceuticals/eudralex/homev2.htm#2c
- Aplicações de renovação
http://ec.europa.eu/enterprise/pharmaceuticals/eudralex/homev2.htm#2c
Informações sobre o produto (Seção 1.3)
A seção 1.3 contém informações sobre
- Resumo das características do produto (SPC), rotulagem e folheto informativo (PIL) (Seção 1.3.1)
- Mock-up (Seção 1.3.2)
- Espécime (Seção 1.3.3)
- Consulta com grupos-alvo de pacientes (Seção 1.3.4)
- Informações sobre produtos já aprovados nos Estados Membros (Seção 1.3.5) (quando aplicável)
- Braile (Seção 1.3.6)
Informações sobre os Peritos (Seção 1.4)
Nesta seção é necessário incluir informações relativas aos especialistas com o fornecimento de relatórios detalhados dos documentos e informações que constituem os Módulos 3, 4 e 5, de acordo com o artigo 12 da Diretiva 2001/83 / CE. Para parte adicional desta seção pode ser utilizado um relatório pericial assinado para as diferentes partes científicas do dossiê. Os requisitos dos Relatórios de Peritos assinados são apresentados abaixo:
- O Resumo Geral da Qualidade, Resumo não-clínico / Resumo e Resumo Clínico / Resumo no Módulo 2,
- Uma declaração assinada pelos especialistas no Módulo 1.4.
- Uma breve informação sobre os antecedentes educacionais, treinamento e experiência ocupacional no Módulo 1.4.
A(s) declaração(ões) do(s) especialista(s) relevante(s) deve(m) ser fornecida(s) para pedidos de pós-autorização.
Nos casos em que os titulares de autorização de comercialização desejam distinguir tal declaração de qualquer
declarações anteriores, o número do procedimento relevante do estado-membro de referência/EMEA
podem ser incluídas no topo.
Subseções de Informações sobre os Peritos apresentadas abaixo:
- Qualidade (Seção 1.4.1)
- Não-Clínico (1.4.2)
- Clínica (Seção1.4.3)
Para maiores informações e modelos relevantes, consulte o Artigo 12 e de acordo com o Anexo I, Parte I 1.4 da Diretiva 2001/83/CE.
Requisitos específicos para diferentes tipos de aplicações (Seção 1.5)
Informações para Aplicações Bibliográficas (Seção 1.5.1)
Com base no artigo 10a da Diretiva 2001/83/CE, os requerentes devem fornecer um documento conciso, resumindo os fundamentos e evidências utilizadas para demonstrar que o(s) componente(s) do medicamento têm um uso bem estabelecido, levando em consideração o nível aceitável de segurança e eficácia, conforme delineado na Parte II.1 do Anexo I da Diretiva 2001/83/CE.
Informações para aplicações genéricas, 'híbridas' ou biossimilares (Seção 1.5.2)
Com base no artigo 10(1), 10(3) ou 10(4) da Diretiva 2001/83/CE, os requerentes devem fornecer aqui um documento conciso, resumindo os fundamentos e provas utilizadas para demonstrar o tipo de medicamento para o qual um pedido é apresentado. Os medicamentos podem ser genéricos, produtos híbridos e biosimilares.
(Estendido) Dados / Exclusividade do mercado (Seção 1.5.3)
A seção 1.5.3 é necessária quando o titular/aplicante da MA desejar reivindicar dados (adicionais) / exclusividade de mercado enquanto solicita uma nova indicação ou alteração na classificação. Neste caso, as disposições e exigências legais relevantes devem ser consideradas.
Circunstâncias Excepcionais (Seção 1.5.4)
De acordo com o artigo 22 da Diretiva 2001/83/CE e o artigo 14(7) do Regulamento (CE) nº 726/2004, uma autorização pode ser concedida em caso de circunstâncias excepcionais, quando procedimentos específicos, em particular relativos à segurança do medicamento, notificação às autoridades competentes de qualquer incidente relacionado à sua utilização são introduzidos pelo requerente. Somente para exceções objetivas e verificáveis, uma autorização pode ser concedida.
Autorização de Comercialização Condicional (Seção 1.5.5)
A seção mencionada é aplicável para procedimento centralizado. As referências para esta seção são o Artigo 14(7) do Regulamento (EC) No 726/2004 e "Diretrizes sobre a aplicação científica e as disposições práticas sobre a Autorização Condicional de Comercialização”.
Avaliação de risco ambiental (Seção 1.6)
De acordo com o Artigo 8 (ca) e (g) da Diretiva 2001/83/CE, qualquer risco potencial do medicamento para o meio ambiente deve ser considerado pelo requerente durante o pedido de autorização de comercialização. As exigências da Diretiva estão relacionadas ao uso, armazenamento e descarte de medicamentos e não são aplicáveis à síntese ou fabricação do produto. Os pedidos de autorização de comercialização de produtos medicinais que não contenham OGM (seção 1.6.1 Não-OGM) e que contenham OGM (GMO 1.6.2) devem incluir uma indicação de quaisquer riscos potenciais no Módulo 1 do dossiê regulamentar.
Para maiores informações, favor consultar "Diretriz sobre a Avaliação de Risco Ambiental para produtos medicinais para uso humano”
Informações relacionadas à Exclusividade do Mercado de Órfãos (Seção 1.7)
Esta seção é aplicável apenas aos medicamentos órfãos. As informações requeridas sobre detalhes e procedimentos estão presentes em "Diretriz da Comissão Européia sobre aspectos da aplicação do artigo 8º do Regulamento (CE) nº 141/2000: Avaliação da similaridade e/ou superioridade clínica de medicamentos órfãos ao avaliar pedidos de autorização de introdução no mercado e variações.”
Seção 1.7.1 Similaridade e Seção 1.7.2 Exclusividade de mercado são aplicáveis, se o medicamento órfão tiver sido autorizado para a condição que cobre a indicação terapêutica proposta que está sendo solicitada, e um período de exclusividade de mercado estiver em vigor, os requerentes devem fornecer um relatório crítico abordando a possível similaridade com o medicamento órfão autorizado e concluindo sobre similaridade ou "não" similaridade (1.7.1). Se o medicamento, que é objeto do pedido de autorização de comercialização, for considerado "similar" a um medicamento órfão abrangido pelas disposições de exclusividade de mercado acima mencionadas, o requerente deve, além disso, justificar que uma das derrogações estabelecidas conforme descrito no artigo 8.3, alíneas (a) a (c) do Regulamento (CE) nº 141/2000(1.7.2).
Information relating to Farmacovigilância (Seção 1.8)
Sistema de farmacovigilância (Seção 1.8.1)
A farmacovigilância é a ciência e as atividades relacionadas à detecção, avaliação, compreensão e prevenção de efeitos adversos ou qualquer outro problema relacionado a drogas.
Isto se aplica durante todo o ciclo de vida da medicina, tanto na fase de pré-aprovação quanto na fase de pós-aprovação. O sistema de farmacovigilância é uma seção muito importante para a aplicação da autorização de comercialização.
A descrição detalhada do sistema de farmacovigilância deve ser fornecida de acordo com o Artigo 8 (ia) da Diretiva 2001/83/CE. A descrição deve incluir prova de que o requerente possui os serviços de uma pessoa qualificada responsável pela farmacovigilância.
A descrição do sistema de farmacovigilância do titular da autorização de comercialização deve seguir as exigências e o formato conforme detalhado no Volume 9A da EudraLex.
Sistema de gerenciamento de risco (Seção 1.8.2)
Detailed description of risk-management system must be provided according to Article 8 (ia) of Directive 2001/83/EC. The detailed description of a risk management system should be provided in the form of an EU Risk Management Plan (EU-RMP), as outlined in Volume 9A of EudraLex.
Informações relacionadas a Ensaios Clínicos (Seção 1.9)
A seção de 1.9 deve ser preparada de acordo com o Artigo 8 (ib) da Diretiva 2001/83/CE para relatar que os ensaios clínicos realizados fora da União Européia cumprem as exigências éticas da Diretiva 2001/20/CE, quando aplicável.
Esta seção deve ser fornecida para todos os novos pedidos (incluindo pedidos de extensão) e outros procedimentos regulatórios relevantes pós-autorização (por exemplo, variações) para os quais os ensaios clínicos apresentaram relatórios.
Informações relativas à Pediatria (Seção 1.10)
De acordo com os artigos 7, 8 e 30 do Regulamento (CE) nº 1901/2006 ("regulamento pediátrico") e o artigo 23 do Regulamento (CE) nº 1901/2006 ("regulamento pediátrico"), esta seção é exigida:
- Para todas as novas aplicações*, para um produto medicinal não autorizado na AEA
- Para pedidos* de novas indicações, novas formas farmacêuticas e novas vias de administração, para medicamentos autorizados que são protegidos ou por um certificado de proteção suplementar, ou por uma patente que se qualifica para a concessão de tal certificado.
- Para Aplicações de Autorização de Comercialização para Uso Pediátrico (PUMA)
*exceto para aplicações genéricas, híbridas, bio-similares e de uso bem estabelecido e medicamentos tradicionais à base de ervas ou homeopáticos
Para o Módulo 1 pode ser fornecido:
Respostas às perguntas nos casos em que os candidatos são aconselhados a incluir nesta seção um documento que lista as perguntas com a resposta do texto narrativo correspondente para cada pergunta, e quando as respostas também contêm dados/documentos novos ou atualizados relacionados aos Módulos 3, 4 e/ou 5. Tais dados/documentos devem ser colocados nas seções relevantes desses Módulos.
Dados adicionais. Esta seção é necessária com base no procedimento de autorização. Dados adicionais podem precisar ser fornecidos como parte de um pedido nacional, descentralizado ou de reconhecimento mútuo. Se tais dados estiverem relacionados aos Módulos 2, 3, 4 e/ou 5, os documentos também deverão ser colocados nas seções relevantes desses Módulos. Os requisitos específicos dos Estados membros para dados adicionais podem ser encontrados no site da Comissão Européia.
CTD-Módulo 2
A introdução geral ao produto medicinal fornecida no Módulo 2 do dossiê CTD, que é harmonizado para todas as regiões (Conselho Internacional para Harmonização de Requisitos Técnicos para Produtos Farmacêuticos para Uso Humano (ICH)). Este módulo é apresentado por documentos resumidos para cada um dos próximos módulos: dados de qualidade, relatórios de estudos não clínicos e clínicos.
CTD-Módulo 3
Module 3 was identified by ICH as the Quality Module. Thus, “Quality” became the global term for CMC (química, manufacturing and controls). A CMC não deve ser confundida com os elementos de controle de qualidade, garantia de qualidade, SOPs (procedimentos operacionais padrão), documentos internos da empresa (especificações, registros de lote, etc.)
Módulo 3 seção também harmonizada para todas as regiões com o fornecimento de informações químicas-farmacêuticas e biológicas para substâncias ativas químicas e produtos medicinais biológicos.
Durante todo o desenvolvimento e após um produto ser aprovado pelas autoridades sanitárias, os aspectos de química, fabricação e controle (CMC) continuam a evoluir e a mudar.
Qualidade Farmacêutica = CMC
QUEMISTRIA
Descoberta de novas entidades/moléculas químicas, purificação de ingredientes ativos (substância medicamentosa), testes analíticos de matérias-primas e produtos acabados.
MANUFATURA
Fabricação em escala de laboratório de substâncias e produtos farmacêuticos, fabricação de suprimentos clínicos para estudos clínicos, escalonamento até o tamanho comercial do lote, produto comercial.
E CONTROLES
Desenvolver especificações/controles apropriados para substâncias e produtos farmacêuticos para garantir a segurança, eficácia e qualidade.
Todas as regiões requerem dossiê de Qualidade CMC/Pharmaceutical
- Iniciar e conduzir ensaios clínicos,
- Para apresentar um novo dossiê de autorização de comercialização (MAA, NDA, BLA, etc.)
- Para atender às exigências regulatórias para a gestão do ciclo de vida e mudanças pós-autorização do produto
Este módulo inclui todo o pacote de documentos relativos a substância farmacêutica (DS) e produto farmacêutico (DP).
Substância de drogas (DS)
A seção de substância farmacêutica é apresentada por sua descrição, incluindo características físicas, químicas ou biológicas, o nome e endereço do fabricante do DS, os limites aceitáveis e os métodos analíticos usados para assegurar a identidade, força, qualidade e pureza da substância farmacêutica.
Também estão incluídas informações para apoiar a estabilidade da substância farmacêutica durante os estudos toxicológicos e o estudo clínico proposto.
PRODUTO FARMACÊUTICO (DP)
Uma lista de todos os componentes, que pode incluir alternativas razoáveis para compostos inativos, usados na fabricação do produto farmacêutico, incluindo tanto os componentes destinados a aparecer no produto farmacêutico quanto os que podem não aparecer, mas que são usados no processo de fabricação são descritos nesta seção.
Abaixo estão listadas as principais informações do produto farmacêutico que devem ser incluídas no dossiê regulamentar:
- A composição quantitativa do medicamento (investigacional ou comercial), incluindo quaisquer variações razoáveis.
- O nome e endereço do fabricante do produto farmacêutico.
- Descrição dos procedimentos de fabricação e embalagem.
- Os limites aceitáveis e métodos analíticos usados para assegurar a identidade, força, qualidade e pureza do produto farmacêutico.
CTD-Módulo 4
Relatórios de estudos não-clínicos
A seção relevante, a localização apropriada para os dados individuais dos animais, está no relatório do estudo no Documento Técnico Comum para solicitações que serão submetidas às Autoridades Reguladoras. O módulo 4 está harmonizado para os EUA e UE com base nos princípios da ICH, e contém todas as seções e subseções necessárias para os relatórios de estudo. Referências às diretrizes da ICH Documento Técnico Comum (CTD).
CTD-Módulo 5
Relatórios de Estudos Clínicos
Módulo 5, esta é a estrutura e o conteúdo dos relatórios de estudos clínicos. Esta parte do CTD apresentou relatórios de estudos clínicos/ humanos, outros dados clínicos e referências dentro de um Documento Técnico Comum (CTD) para registro de um produto farmacêutico para uso humano. Estes elementos devem facilitar a preparação e a revisão de um pedido de comercialização. O módulo 5 está harmonizado para os EUA e UE com base nos princípios da ICH, e contém todas as seções e subseções necessárias para relatórios de estudo. Referências às diretrizes da UE Documento Técnico Comum (CTD).
Precisa de ajuda para preparar um dossiê regulamentar? Ver e consultar redatores freelance de regulamentação em Kolabtree.
Referências adicionais
- https://www.ich.org/page/ctd
- https://www.fda.gov/media/128163/download
- https://www.fda.gov/media/135573/download
- https://ec.europa.eu/health/sites/default/files/files/eudralex/vol-2/b/update_200805/ctd_05-2008_en.pdf
- https://ec.europa.eu/health/sites/default/files/files/eudralex/vol-1/dir_2001_83_consol_2012/dir_2001_83_cons_2012_en.pdf
- https://eur-lex.europa.eu/legal-content/EN/TXT/PDF/?uri=CELEX:02001L0083-20121116&from=DE
- https://eur-lex.europa.eu/legal-content/EN/TXT/PDF/?uri=CELEX:02004R0726-20130605&from=EN
- https://ec.europa.eu/health/sites/default/files/files/eudralex/vol-1/reg_2000_141_cons-2009-07/reg_2000_141_cons-2009-07_en.pdf
- https://www.ema.europa.eu/en/human-regulatory/research-development/clinical-trials-human-medicines
- https://eur-lex.europa.eu/legal-content/EN/TXT/?uri=celex%3A32006R1901
- https://clinicaltrials.gov/ct2/home
Kolabtree helps businesses worldwide hire freelance scientists and industry experts on demand. Our freelancers have helped companies publish research papers, develop products, analyze data, and more. It only takes a minute to tell us what you need done and get quotes from experts for free.

Unlock Corporate Benefits
• Secure Payment Assistance
• Onboarding Support
• Dedicated Account Manager
Sign up with your professional email to avail special advances offered against purchase orders, seamless multi-channel payments, and extended support for agreements.


