



Shrinidh Joshi, medical device expert on Kolabtree, provides a comprehensive guide to medical device design, design controls, validation & verification, regulatory requirements and risk management.
In the previous article, we took a look at the overview of the medical device development process from the ideation to the discovery phase. In this article, we will focus more on medical device design, design controls, and compliance.
Medical Device Design: IEC & ISO regulations and compliance
By now you know that for market entry your medical device needs to qualify certain regulatory requirements and standards. Medical device standards such as the International Electrotechnical Commission (IEC) or the International Organization for Standardization (ISO) allow medical device manufacturers, designers, laboratories and all other medical device development service providers such as CDMO to inspect, assess and maintain their devices and equipment to certain quality and usability standards.
The (IEC published the first of its kind medical devices standard in 1970, IEC 60601-1. IEC 60601-1, Medical electrical equipment – Part 1: This is the internationally recognized standard that addresses general requirements for medical electrical equipment and devices covering standards for basic safety and essential performance [4].
The IEC 60601-1 document has been revised periodically to stay in alignment with the latest medical development and technological breakthroughs in the medical device field. Most recent change came into fruition in 2012 (Amendment 1 to IEC 60601-1). This revised standards contains the requirements for human factor consideration, essential performance evaluation of medical devices, usability and commands. It also includes software as a medical device and specifies its adoption of a formal development life cycle. Also included in the scope of revised IEC 60601-1 are newer and revised technical specifications for hazards (both electrical and mechanical), medical device labeling requirements (including new labeling standards) and documentation.
Medical Device Design: ISO Standards
The International Organization for Standardization also has specifications for medical device standards. ISO 13485 and ISO 14971 are widely used standards across the world for medical device quality management. Other than these international standards, certain standards are region-specific and all of them are adopted from international standards with little modification and limitation.
If a medical device company is manufacturing or selling medical devices in the US, the medical device will be regulated by the FDA. American National Standards Institute (ANSI) is the representative of ISO standards in the US.
There are two more similar organizations: the Association for the Advancement of Medical Instrumentation (AAMI) and the American Society for Quality (ASQ) that defines standards for the US.
If a medical device company had designed a device considering ISO standards, there is also a possibility that the FDA may not approve the device. As FDA has its own set of procedures for risk management derived from both international and regional standards, which includes:
(international standard.)
- ANSI/AAMI/ISO 14971:2007 (R2010), Medical devices – Application of risk management to medical devices (A regional standard with additions and modifications from the referred international standard.) [5].
In the case of quality management standard, it does not follow the international or regional version of the ISO 13485 standard. This is because the FDA has different guidelines for quality management in medical devices for the US market.
Whereas, if the medical device company is considering the European Union, the European Committee for Standardization (CEN) is the standardization adopted from ISO and the European Committee for Electrotechnical Standardization (CENELEC) is the regional standard inspired by IEC.
CEN is a bit modified as per requirement from ISO and written with an “EN” prefix. E.g.:
- EN ISO 13485:2012, Medical devices — Quality management systems — Requirements for regulatory purposes.
- EN ISO 14971:2012, Medical devices — Application of risk management to medical devices
National members adopt these standards from the EU while adding their prefix. For Switzerland, Swiss Standards publishes standard with “SN” as a prefix such as SN EN ISO 13485:2012 and SN EN ISO 14971:2012.
In the case of Canada, the Canadian Standards Authority (CSA) is the representative organization for ISO.
Medical Device Regulations and Design Control
Medical device manufacturers need to follow Design Control guidelines since the regulatory bodies like FDA, European Commission, Health Canada, and others want to ensure that the medical devices are safe for potential users before manufacturers start to market the devices. As I have mentioned in the above section, the FDA doesn’t follow ISO 13485 as it has different requirements for quality management. Design controls are defined under FDA 21 CFR 820.30 which has a similar intent to section 7.3 Design and Development described under the guidelines for ISO 13485. Additionally, FDA incorporates Current Good Manufacturing Practice (cGMP) requirements into the quality system regulation to follow good quality practices for medical device designs [6].
The regulation provides a framework to implement the design control to a wide variety of devices. The framework delivers flexibility for both regulatory compliances as well as the internal design and development process.
To successfully implement design control of medical devices, professionals with both technical and non-technical backgrounds, such as business administration, life science, engineering, computer science, and the arts are required.
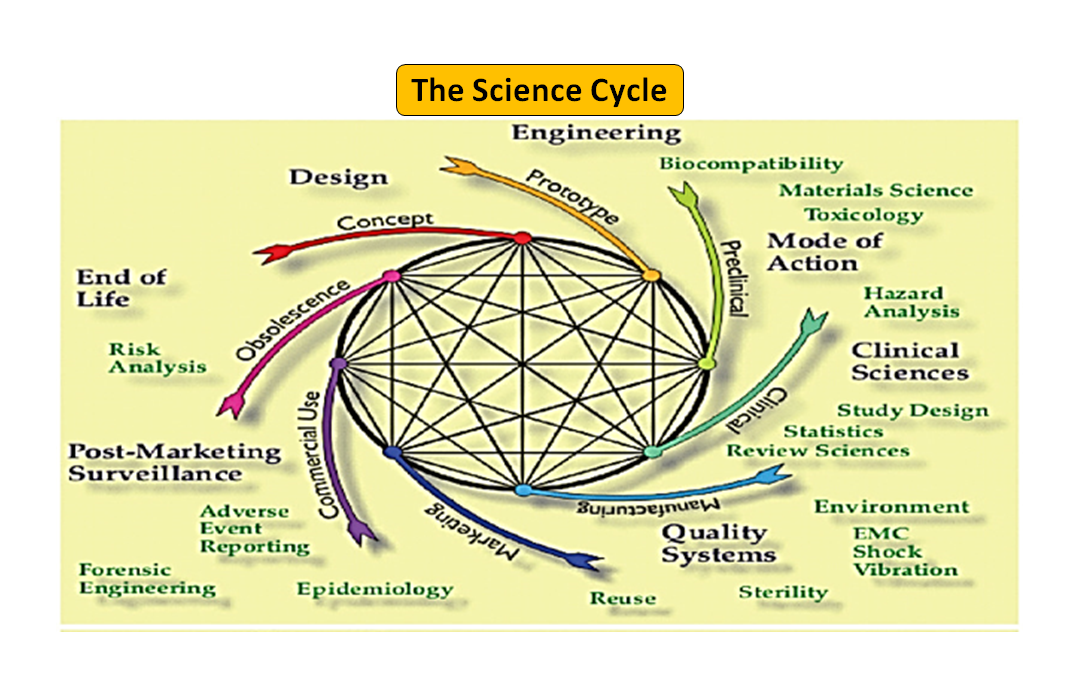
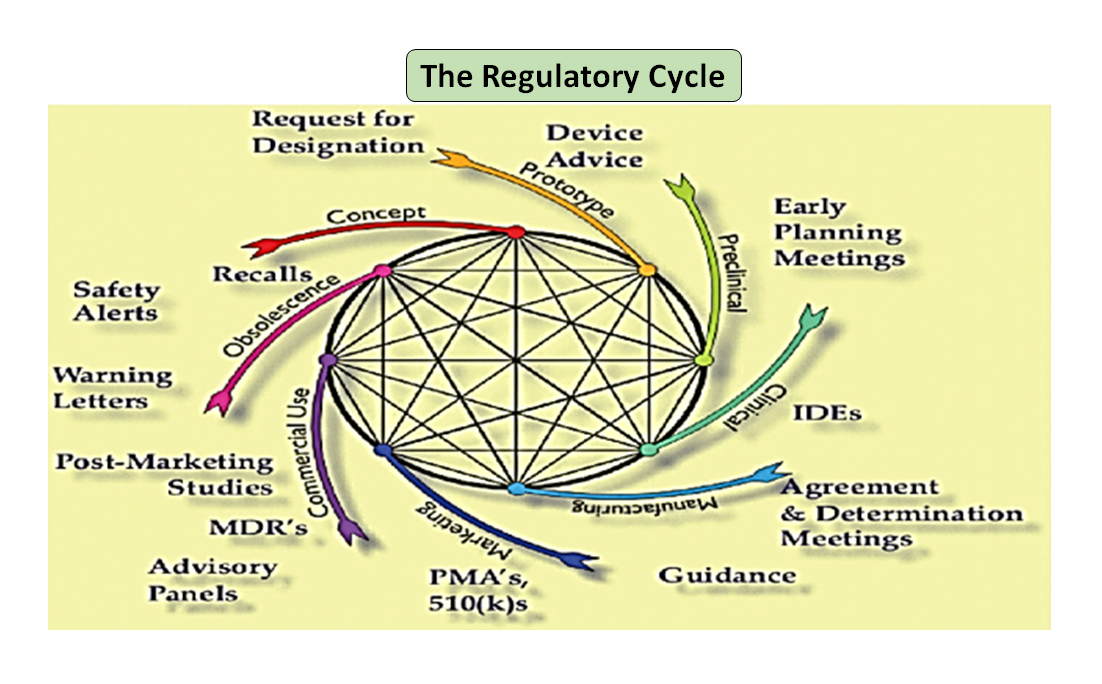
Figure 1: Total product life cycle. The science cycle and the regulatory cycle (Adapted from [7]).
It is noteworthy that the medical device life-cycle starting from innovation to regulatory approval and commercialization is a series of interconnected steps which drives the device development (see Figure 1: Total product cycle). In the beginning, prototypes designed by your engineers are bench tested to optimize the design, tested for biocompatibility, extractables, leachables, flexibility or overall strength of your device. Role of your company’s regulatory advisor is to browse through the regulatory database to suggest to you a guidance document that can help you determine if your product will be regulated as a medical device or not. Intended use of your medical device and its mode of operation or action will guide you in the device design and will also decide your regulatory pathway whether 510(k), PMA, De Novo, Pre-sub, IDE, HDE, master files etc.
As illustrated in Figure 1, both the scientific and regulatory processes are intertwined throughout the product life cycle. Just as different parts of the science life cycle are interconnected, the science and the regulatory requirements are intertwined, each informing and determining the other. There is an opportunity to build connections, both at the FDA and in manufacturers, so parts of the life cycle do not risk only being considered in isolation. For example, it is not uncommon for a premarket application to be reviewed without considering the postmarket experience of similar products.
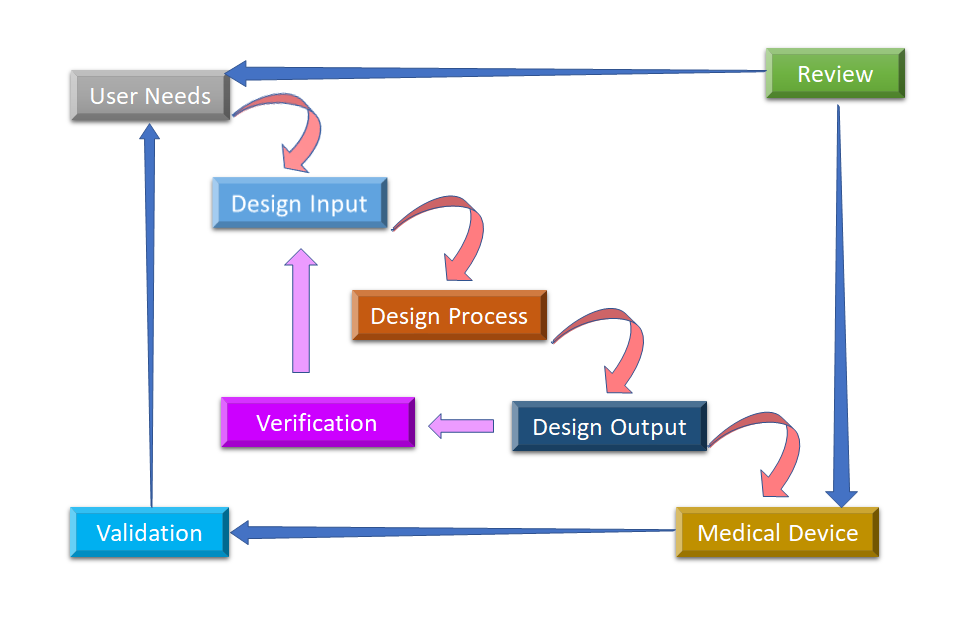
Figure 2: Waterflow design process for medical device design control (Adapted from [8]).
The initial phase from which Design Control starts is Design Input development and approval, which consists of device design and manufacturing processes to be carried out in the production phase. Design control is a holistic approach and doesn’t end with transferring the design to the production phase, once the design is finalized. It also impacts manufacturing processes according to the changes in the design phase or even post-production feedback. It is an ongoing process to develop a product that is usable for a user and thus for the enhanced product, it considers revolutionary changes from usage patterns as well as analyzing failed products. You can see in Figure 2, how Design Control can be performed in the waterfall design process.
- User Needs:- Requirements are defined considering the market need and the device is designed to address that need. After a series of evolution, the medical device design is finalized and transferred to production for manufacturing. There is a need for feedback during every step of this process.
- Design Input: This is an iterative process. When an organization decides to address the particular need, they review and test the acceptability of design input derived from the need. At that point, the iterative process of converting requirements into device design starts.
- Design Process: These design inputs are converted into design output by converting those requirements into high-level specifications (which are Design Output).
- Design Output: The verification process confirms whether the specifications are satisfying requirements or not. And the output becomes the input to revise the requirements and this process goes on until Design Output is aligned with the Design Input.
- Medical Device: Once the final design is ready, it is transmitted to the production facility for mass manufacturing. Design control regulation mandates Design History File (DHF), which illustrates the linkages and relationships between all the Design Controls and help to trace all changes throughout the entire product development process.
Medical device companies can take a paper-based or a software-based approach, especially developed for Design Control; design history files must be traceable as well as accessible to all the team members.
The flow chart below shows a case study for medical device design control.

Medical Device Design: Why Traceability is Important
Currently in the realm of the medical devices industry, it is an ideal practice to develop a traceability matrix that can illustrate the links and relations between user needs, design inputs and outputs, design verification and validation. When you are in the early phase for your device development you can maintain the device traceability using a spreadsheet or document version but as you move forward, its good idea to use cloud-based project management and document sharing platforms such as Microsoft Teams, Asana, Trello or whichever platform is suitable for your organization. The goal is as your project progresses you need to find an option which can save time because the old-school method of maintaining a traceability matrix might consume a lot of your time which you should rather be focusing on design verification and validation.
A Design Control traceability matrix is vital to product development teams, and especially for project managers, because traceability shows the relationship and linkages between all of the Design Controls. How do User Needs relate to Design Inputs? How do Design Outputs relate to Design Inputs? How do Design Verifications link to Design Inputs and Design Outputs? How do Design Validations relate to User Needs? A traceability matrix is an invaluable tool to show a high-level view and the flow of medical device product development from beginning to end.
Best practice product developers have relied on Design Controls traceability for many, many years. And now ISO 13485:2016 also makes traceability a requirement. As cited in ISO 13485:2016, 7.1 Planning of Product Realization, 1. c) required verification, validation, monitoring, measurement, inspection and test, handling, storage, distribution and traceability activities specific to the product together with the criteria for product acceptance; And 7.3.2 Design and Development Planning, 1. e) the methods to ensure traceability of design and development outputs to design and development inputs [9].
Medical Device Design: Verification and Validation
Every medical device must meet the functionality, usability, and reliability objectives to get a successful share in the market.
In addition, your stakeholders (patients, prescribers, regulators or end-users) will also pay attention to the safety and effectiveness of your device. It is very likely that your device is designed to address an unmet need which can be critical to life for example a ventilator or a diagnostic device which can detect heart disease. Therefore, iterative testing of your device with verification and validation is crucial. These two steps in the design process are aimed to confirm that your medical device is in alignment with the requirements of users and it is performing as per its intended use. In simple terms, design verification and validation can ensure that your device is actually doing what it is supposed to be doing. Design verification and validation are also to ensure regulatory requirements, standards, product quality, and manufacturing process of your medical device. Design verification can evaluate if your design output is in compliance with the specified requirements, specifications or regulatory requirements that are specified in the design input. On the other hand, design validation is meant to evaluate whether your medical device is delivering benefits based on the need of the end-users.
Design verification asks: “Did we design the device right?”
Design validation asks: “Did we design the right device?”
Medical devices may consist of different technology shapes, sizes, and different levels of complexity. Verification and validation (V&V) activity is driven by the regulatory environment and must follow international standards. Standardized V&V activities can streamline the manufacturing process as well as enhance the approval process. Additionally, automated testing, diagnostic techniques, and data collection tools can enhance the V&V process [10].
- Product validation vs. Process validation
- Medical device design/product validation:- Conforming to the user and patient needs, i.e. Does the device work right?
- Process validation:- Manufacturing process meets predetermined specifications.
Noteworthy to remember is Design/Product validation ≠ process validation. Regulatory agencies require both design/product validation and process validation individually, so both of them need to be equally taken into account during regulatory submission.
How early in the development process should we think about validation? A medical device company should understand that it is never too early to start validation work, a company should start validating sooner than later to figure out that it is going on the right path and solving the right problem.
Validation (also V&V) being an iterative process consumes a good investment, when planned poorly. A strongly-defined test strategy can help you optimize cost as well as the test period to make the product market-ready on time.
The complexity of any testing strategy depends on technologies to be used and geographical target markets. The test strategy should cover at least six parameters mentioned below:
- Targeted geographies and associated standards;
- Time to market;
- A standard to be followed with a version;
- Testing Labs – internal or independent labs;
- Defining the sequence of tests;
- Presenting the test result
Accordingly, tests used for the verification and validation process also need to be validated. This is to ensure that you measure what you need to measure because a wrong test will deliver wrong outputs of usability and functionality. Medical device companies need an effective and well documented V&V, which complies with associated regulations.
Medical Device Design: Risk Management
Risk migration strategy vs. Risk management plan
Risk management procedures for medical devices are enforced under internationally accepted compliance standard ISO 14971:2007 Medical Devices – “Application of Risk Management to Medical Devices”. Apart from this, risk management policies need to be incorporated across all the stages of medical device design and development and should be also associated with design control aspects as well [10].
Risk management never ends (at least in theory!). The philosophy of risk management is that it should not be about a hard and fast set of rules. Risk management and risk migration strategy are about understanding the intent of risk management and approaching the process logically and systematically. In other words, don’t just follow the rules…think!
Considering the complexity of medical device design, focused risk management practices help ensure usability, safety, and regulatory compliance. It is a process of identifying, controlling, and preventing the failure that may cause hazards to users. It also mandates identifying associated risks.
Figure 3 shows all the steps involved in the risk management process. The process starts with the identification of hazards and then associated risk is measured based on the consequences of hazards and their possibility of risk.
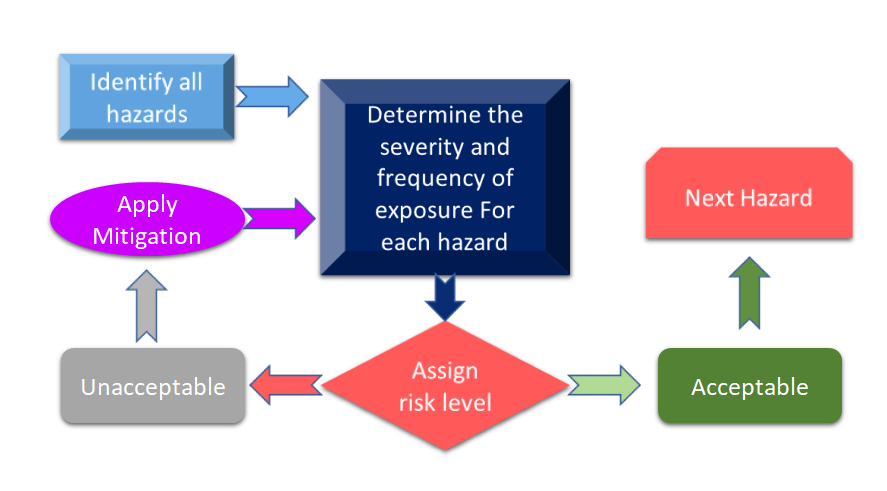
Figure 3: Risk management process for Medical Device (Adapted from [11]).
In case if the risk identified during the risk management process for medical devices is above the defined criteria, then you will require to have a risk mitigation. The level of risk is dependent on several parameters including but not limited to your device, technologies, and in some cases to the way your company is handling the risk mitigation process. It is always advisable to conduct hazard analysis for your device to see what standards can be applied to your device. In the recent revision of ISO 14971: International Standard for Risk Management of Medical Devices, risk analysis and Preliminary Hazard Analysis (PHA) are identified as prime requirements for your medical device [12]. In a simplified manner PHA is meant to provide the initial framework for risk assessments and management and PHA covers both risk analysis and risk evaluation. As per definition, PHA comprises a list of hazards, harms, any hazardous situations, formulated from your devices’ materials of construction (MoC), components or raw material used in your devie, and human-device or manual interfaces, use environment, operating principle, and other relevant factors [13].
Conclusion
In the end, for every medical device startup or a well-established organization, it is important to remember that reading the regulations gains you nothing but understanding the philosophy buys you a lot!
Bottom line: when comes to risk analysis and planning:
- Should be utilized early and throughout the design and development process,
- Often generated new information to feedback into the design and development process (à current/future devices),
- No amount of planning can eliminate all hazards and risks…but you can mitigate many of them! (Following the design control philosophies described here automatically mitigates risk!)
Every medical device’s route to market is complex due to the various factors to be taken into consideration such as usage patterns, material, user experience, regulations, and more.
Need help with medical device design? Browse experienced medtech industry experts on Kolabtree or post your project for free to get proposals.
REFERENCES AND RESOURCES
- https://www.welldoc.com/health-plans/
- https://ec.europa.eu/docsroom/documents/10337/attachments/1/translations
- FDA, 2005, Total Product Lifecycle, FDA-CDRH Presentation by CDRH Director Dr. David Feigal, http://www.fda.gov/cdrh/strategic/presentations/ tplc.html
- Pietzsch, Jan & Shluzas, Lauren & Paté-Cornell, Marie-Elisabeth & Yock, Paul & Linehan, John. (2009). Stage-Gate Process for the Development of Medical Devices. Journal of Medical Devices. 3(2).
- Regulatory Strategies for the Third Edition of IEC 60601-1 Retrieved 9 September 2020.
- https://www.meddeviceonline.com/doc/an-introduction-to-international-medical-device-standards-0001
- https://www.fda.gov/files/drugs/published/Design-Controls—Devices.pdf
- Feigal DW. Appendix D. Impact of the Regulatory Framework on Medical Device Development and Innovation. Institute of Medicine (US) Committee on the Public Health Effectiveness of the FDA 510(k) Clearance Process; Wizemann T, editor. Public Health Effectiveness of the FDA 510(k) Clearance Process: Balancing Patient Safety and Innovation: Workshop Report. Washington (DC): National Academies Press (US); 2010. Appendix D, Impact of the Regulatory Framework on Medical Device Development and Innovation. Available from: https://www.ncbi.nlm.nih.gov/books/NBK209794/.
- 1997, FDA CDRH 1997, Design Control Guidance for Medical Device Manufacturers
- https://starfishmedical.com/blog/iso-134852016-section-7/?doing_wp_cron=1599995964.4528369903564453125000
- Teixeira, M. B., and Bradley, R., 2003, Design Controls for the Medical Device Industry, Marcel Dekker, New York.
- ISO 14971:2019 – Medical devices – Application of risk management to medical devices
- ISO/TR 24971:2020 – Medical devices – Guidance on the application of ISO 14971
All articles in this series:
Medical Device Development and Design: A Definitive Guide
Medical Device Development: 3 Tips for Success
Medical Device Design: The Essential, Step-by-Step Guide
Medical Device Commercialization: 9 Steps from Sketch to Launch
How to Overcome Medical Device Commercialization Challenges
Medical Device Launch: Key Steps to Bring Your Product to Market
Medical Device Post-Market Surveillance: A Comprehensive Guide
Kolabtree helps businesses worldwide hire freelance scientists and industry experts on demand. Our freelancers have helped companies publish research papers, develop products, analyze data, and more. It only takes a minute to tell us what you need done and get quotes from experts for free.

Unlock Corporate Benefits
• Secure Payment Assistance
• Onboarding Support
• Dedicated Account Manager
Sign up with your professional email to avail special advances offered against purchase orders, seamless multi-channel payments, and extended support for agreements.


