



Nare Simonyan, freelance regulatory affairs specialist のKolabtreeでは、規制当局の書類とそのフォーマットに関する包括的なガイドを提供しています。
はじめにレギュラトリー・ドシエとは?
Regulatory dossier is a package of documents, which may include all required information regarding newly developed drug products and/or generics, which is required by EU and US regulatory authorities for granting マーケティング authorization approvals. The main information that is included in the package is administrative information, data related to the quality, safety and efficacy of drug product, which can be submitted by CTD (Common Technical Document) format both paper and electronic version. Since the information submitted in paper format was enormous, agencies are now encouraging applications to be submitted in eCTD format.
CTD(コモン・テクニカル・ドキュメント
2003年、ICH(International Council of Harmonization)のメンバーは、すべての品質、安全性、有効性に関する情報を、かつてCTDと呼ばれていた共通のフォーマットにまとめることに合意しました。CTDは、NDA(New Drug Application)、MAA(Marketing Authorization Application)、およびグローバルメディシナルアプリケーションのモジュール1から5までのフォーマット/構造である。モジュール1には、国ごとに異なる行政上の地域情報が含まれています。モジュール2、3、4、5はすべての地域で共通です。CTDの構造は、治験申請と商業申請の両方(IND(治験薬)とNDA(米国)、IMPD(治験薬)とMAA(EU)、およびグローバル申請)に適用されます。
The CTD was primarily used for new marketing applications such as NDA, BLA (Biologics License Application), MAA, NDS (New Drug Substance), JNDA (Japanese New Drug Application), etc. With additional guidance and guidelines from the FDA and EMA, the CTD is now required for all applications, including those for 臨床試験—IMPD and INDs. All Drug Master Files (DMF) and Active Substance Master Files (ASMF) must follow the structure of the CTD. The process of submitting regulatory dossier is regulated by Code of Federal Regulations (CFR) (Law in US) and Directives (Law in EU).
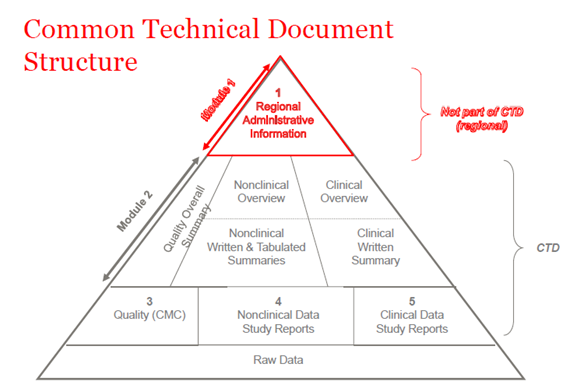
図1.CTDの構造
CTD-Module 1
USモジュール1の仕様:管理情報と処方情報
e-CTDモジュール1セクションには、管理用およびラベル用の文書が含まれます。すべての申請書および関連する提出物は、モジュール1の文書に対して同じ組織構造を持っています。
モジュール1で提供されるドキュメントの仕様は以下の通りです。
カバーレター(セクション1.2
カバーレターには、審査過程でのコミュニケーションに役立つ適切な情報が含まれています。カバーレターには以下の情報を含めることが推奨されています(詳細は "Electronic Common Technical Document (eCTD) v4.0 技術的適合性ガイド")
- 適切な規制情報、および提出された情報への希望するハイパーリンクを含む、提出物の規制に関する記述
- 提出物のおおよそのサイズ(例:2ギガバイト)を含む、提出物の技術的な説明
- ファイルのウイルスチェックに使用したソフトウェア(名称、バージョン、会社名)の説明とともに、提出物がウイルスに感染していないことを表明すること
- 提出物に関する規制上および技術上の連絡先(電子メールアドレスを含む)。
フィールドコピー認証(1.3項
Field Copy Certificationは、市販申請用のeCTDに含まれるべきものです。eCTD を FDA に提出することを通知する手紙を地区事務所に送ることができます。手紙には以下が含まれるべきである。
- 薬剤およびアプリケーション番号
- FDAセンター&ディビジョン
- アプリケーションはeCTD形式です。
参考文献(1.4項
参考資料には、以下のサブセクションが含まれます(追加コンテンツも必要な場合があります)。
- 許可証(LOA)の発行
- 過去に提出したeCTDフォーマット以外の情報の相互参照について
情報の修正(1.11項
これは、提出される情報がモジュール2、3、4、5のどの見出しにも当てはまらない場合に、情報要求(IR)に対する回答を提出する際に使用することができます。IRの回答がモジュール2~5で提出された文書に影響を与える場合は、新規または差し替えの文書をモジュール2~5の適切な場所に提出し、セクション1.11の文書で参照する必要があります。
マーケティング・アニュアルレポート(1.13項
ポスト・マーケティング・リクエスト/コミットメント・ファイルに記載されている各研究または試験については、ブックマークを含める必要があります。
ラベリング(1.14項
1.14項では、特定のラベリング文書を提供する方法を説明しています。2253申請*のためのドラフト表示、最終表示、上場医薬品表示、治験薬表示、外国語表示、製品表示が含まれる場合があります。提供される情報には、ラベリングの歴史、内容、サンプルが含まれる場合があります。
Form FDA 2253*です。 人体用医薬品および生物学的製剤の広告および販売促進用ラベルの送付について
広告および販促用ラベル素材(セクション1.15
米国では、広告や販促用のラベリング資料が制限されているため、FDAガイダンスに記載されている要件を反映させる必要があります。 電子および非電子形式での規制当局への提出物の提供-ヒト用処方薬の販売促進用ラベルおよび広告資料.
マーケティング後の報告義務の一環として提出される販促資料については、参考文献やラベルへのハイパーテキストリンクを提供することが提案されています。参考文献は審査の効率を高めます。
リスク評価および緩和戦略(REMS)(セクション1.16
REMSサプリメントは、新しいREMSまたは承認されたREMSの修正(メジャーおよびマイナー)を提案することを目的としています。適切なFDAフォーム提出タイプのREMSサプリメントを選択する必要があります。REMS評価、REMS改訂、およびREMS対応はサプリメントではない。この場合 投稿タイプ は、FDAフォームを記入する際に「その他」を選択してください。
欧州連合(EU)モジュール1の仕様:管理情報と処方箋情報
カバーレター(Section 1.0
各申請書にはカバーレターを添付してください。カバーレターの付録として「審査員への注意事項」文書を添付することができます。これは、ナビゲーションを容易にするためにさらなる情報を提供する必要がある場合に備えてのことです。
総合目次(セクション1.1
申請書の種類ごとに包括的な目次を提供する必要があります。この目次には、当該申請書の一部として提出されたすべてのモジュールセクションを含めることができます。新規申請の場合には、すべてのセクションを記載する必要があります。
応募用紙(1.2項
提出物の種類に応じて、関連する申請書を規制当局の書類に含める必要があります。
各種申請書は、欧州委員会のウェブサイト / (英語)でご覧いただけます。
DGエンタープライズ。
- 新しいアプリケーションと拡張アプリケーション http://ec.europa.eu/enterprise/pharmaceuticals/eudralex/homev2.htm#2b
- バリエーションアプリケーション
http://ec.europa.eu/enterprise/pharmaceuticals/eudralex/homev2.htm#2c
- 更新申請
http://ec.europa.eu/enterprise/pharmaceuticals/eudralex/homev2.htm#2c
製品情報(1.3項
セクション1.3には、以下の情報が含まれています。
- 製品特性の概要(SPC)、ラベル、パッケージ・リーフレット(PIL (セクション1.3.1)
- モックアップ (セクション1.3.2)
- 標本 (セクション1.3.3)
- 対象となる患者様グループとの協議 (セクション1.3.4)
- メンバー国で既に承認されている製品情報 (セクション1.3.5) (該当する場合)
- 点字 (セクション1.3.6)
エキスパートに関する情報(1.4項
このセクションには、指令 2001/83 / EC の第 12 条に従ってモジュール 3、4 及び 5 を構成する文書及び情報の詳 細なレポートを提供する専門家に関する情報を含める必要がある。このセクションの追加部分には、書類の異なる科学的部分のための署名入り専門家レポートを使用することができる。署名入り専門家報告書の要件を以下に示す。
- モジュール2の「クオリティー・オーバーオール・サマリー」、「ノンクリニカル・オーバーオール/サマリー」、「クリニカル・オーバーオール/サマリー」。
- モジュール1.4の専門家が署名した宣言書。
- モジュール1.4の教育的背景、トレーニング、職業経験についての簡単な情報。
承認後の申請には、関連する専門家の宣言書を提出しなければならない。
製造販売承認者が、その宣言をいかなるものとも区別したい場合には
過去の申告書、参照加盟国/EMEAの関連手続き番号
はトップに含まれることがあります。
以下、「専門家に関する情報」のサブセクションを紹介します。
- 品質 (1.4.1項)
- ノンクリニカル(1.4.2
- 臨床 (セクション1.4.3)
詳細および関連するテンプレートについては、第 12 条を参照し、指令 2001/83/EC の付属書 I、パート I 1.4 に従ってください。
アプリケーションの種類に応じた固有の要件(1.5項
書誌的応用のための情報(1.5.1項
指令2001/83/ECの第10a条に基づき、申請者は、指令2001/83/ECの附属書IのパートII.1に概説されているように、医薬品の構成要素(複数可)が、安全性と有効性の許容レベルを考慮した上で、確立された用途を有していることを実証するために使用された根拠と証拠を要約した簡潔な文書を提出する必要がある。
ジェネリック、「ハイブリッド」またはバイオシミラーの申請に関する情報(1.5.2項
指令2001/83/ECの第10条(1)、第10条(3)または第10条(4)に基づき、申請者はここで、申請が提出される医薬品の種類を証明するために使用される根拠と証拠を要約した簡潔な文書を提供する必要がある。医薬品には、ジェネリック医薬品、ハイブリッド製品、バイオシミラーなどがあります。
(拡張)データ/市場の独占権(1.5.3項)
1.5.3項は、MA保有者/申請者が、新しい適応症や分類の変更を申請する際に、(追加の)データ/市場独占権を主張したい場合に必要です。この場合には、関連する法律の規定と要件を考慮しなければならない。
例外的状況(1.5.4項
指令2001/83/ECの第22条及び規則(EC)No726/2004の第14条(7)によれば、例外的な状況の場合には、特に医薬品の安全性、その使用に関するあらゆる事故の管轄当局への通知に関する特定の手続きが申請者によって導入されている場合には、認可を受けることができる。客観的で検証可能な例外の場合にのみ、認可を与えることができる。
条件付販売許可(1.5.5項
本項は、集中管理型の手続きに適用されます。このセクションの参考文献は、Regulation (EC) No 726/2004のArticle 14(7)と"条件付販売許可の科学的適用と実務上の取り決めに関するガイドライン".
環境リスクアセスメント(1.6項
指令2001/83/ECの第8条(ca)及び(g)に従い、医薬品の環境に対する潜在的なリスクは、申請者が製造販売承認の承認申請中に考慮しなければならない。この指令の要求事項は、医薬品の使用、保管、廃棄に関するものであり、製品の合成や製造には適用されません。遺伝子組み換え作物を含まない医薬品(Non-GMO Section 1.6.1)および遺伝子組み換え作物を含む医薬品(GMO 1.6.2)の製造販売承認申請には、規制当局用書類のモジュール1に潜在的なリスクの表示を含める必要があります。
詳細については、以下をご参照ください。ヒト用医薬品の環境リスク評価に関するガイドライン"
オーファンマーケットの独占権に関連する情報(第1.7項
このセクションは、オーファン・メディケーションにのみ適用されます。詳細や手続きについての必要な情報は、"欧州委員会は、Regulation (EC) No 141/2000のArticle 8の適用に関する側面についてのガイドラインを作成した。製造販売承認申請およびその変更を評価する際の、希少疾病用医薬品の類似性および/または臨床的優位性の評価。"
1.7.1項の「類似性」と1.7.2項の「市場独占性」が適用される場合、申請される予定の治療適応症をカバーする条件でオーファン医薬品が承認され、市場独占期間が有効であれば、申請者は承認されたオーファン医薬品との類似性の可能性を検討し、類似性または「非」類似性について結論を出す批判的報告書を提出しなければなりません(1.7.1)。製造販売承認申請の対象となる医薬品が、上記の市場独占権規定の対象となるオーファン医薬品と「類似」しているとみなされる場合、申請者はさらに、規則(EC) No 141/2000の第8.3条(a)~(c)項に記載されている軽減措置の1つを正当化する理由を提供しなければならない(1.7.2)。
Information relating to ファーマコビジランス (セクション1.8)
ファーマコビジランスシステム(1.8.1項
ファーマコビジランス(Pharmacovigilance)とは、副作用やその他の薬物関連の問題を検出、評価、理解、予防するための科学と活動のことです。
このことは、医薬品のライフサイクル全体を通して、承認前の段階でも承認後の段階でも同じように適用されます。ファーマコビジランスシステムは、製造販売承認申請のための非常に重要なセクションです。
指令2001/83/ECの第8条(ia)に従い、ファーマコビジランスシステムの詳細な説明を提供しなければならない。説明には、申請者がファーマコビジランスに責任を持つ有資格者のサービスを受けていることの証明を含まなければならない。
製造販売承認者のファーマコビジランスシステムの記述は、EudraLex の Volume 9A に詳述されている要件と形式に従うべきである。
リスク管理体制(1.8.2項
Detailed description of risk-management system must be provided according to Article 8 (ia) of Directive 2001/83/EC. The detailed description of a risk management system should be provided in the form of an EU Risk Management Plan (EU-RMP), as outlined in Volume 9A of EudraLex.
臨床試験に関連する情報(1.9項
1.9 のセクションは、欧州連合外で実施された臨床試験が指令 2001/20/EC の倫理的要件を満たしている旨の報告書を、該当する場合には、指令 2001/83/EC の第 8 条 (ib) に従って作成しなければならない。
このセクションは、臨床試験が報告書を提出したすべての新規申請(延長申請を含む)、およびその他の関連する承認後の規制手続き(例:バリエーション)について提供されるべきである。
小児科に関する情報(1.10項
Regulation (EC) No 1901/2006 ('pediatric regulation') の Article 7, 8 and 30 および Regulation (EC) No 1901/2006 ('pediatric regulation') の Article 23 に従って、このセクションは必要です。
- EEAで認可されていない医薬品のすべての新規申請*について
- 追加保護証明書、またはそのような証明書を付与する資格のある特許によって保護されている認定医薬品の新効能、新剤形、新投与経路の申請*について。
- 小児用医薬品製造販売承認申請(PUMA)について
*ジェネリック、ハイブリッド、バイオシミラー、確立された使用用途、伝統的なハーブやホメオパシーの医薬品を除く
モジュール1の提供が可能です。
質問への回答 申請者がこのセクションに、各質問に対応するナラティブテキストの回答を含む質問をリストアップした文書を含めることを助言され、回答にモジュール3、4、および/または5に関連する新規または更新されたデータ/文書が含まれる場合。このようなデータ/資料は、これらのモジュールの関連セクションに配置する必要があります。
追加データ このセクションは、承認手順に基づいて必要となります。追加データは、国内、分散型、または相互承認申請の一部として提供する必要があるかもしれない。 そのようなデータがモジュール2、3、4及び/又は5に関連する場合、文書はそれらのモジュールの関連セクションにも置かれるべきである。追加データに関する加盟国固有の要求事項は、以下のウェブサイトに掲載されています。 欧州委員会.
CTD-Module 2
医薬品の一般的な紹介は、CTD文書のモジュール2のセクションで提供され、これはすべての地域で調和されています(The International Council for Harmonization of Technical Requirements for Pharmaceuticals for Human Use (ICH))。このモジュールでは、品質データ、非臨床試験および臨床試験報告書という今後の各モジュールの要約文書が提示されています。
CTD-Module 3
Module 3 was identified by ICH as the Quality Module. Thus, “Quality” became the global term for CMC (ケミストリー, manufacturing and controls). CMCは、品質管理、品質保証、SOP(標準作業手順書)、会社の内部文書(仕様書、バッチ記録など)の要素と混同してはいけません。
また、モジュール3のセクションでは、化学的活性物質と生物学的医薬品の化学的・医薬品的・生物学的情報を提供することで、すべての地域で調和を図っています。
開発期間中、そして製品が保健所に承認された後も、化学、製造、管理(CMC)の側面は進化し、変化し続けます。
医薬品の品質=CMC
CHEMISTRY
新規化学物質/分子の発見、有効成分(原薬)の精製、原材料や最終製品の分析試験。
製造業
原薬・製剤のラボスケール製造、臨床試験用消耗品の製造、商用バッチサイズへのスケールアップ、商用製品の製造
とコントロール
安全性、有効性および品質を確保するために、原薬および製剤の適切な仕様/管理を策定する。
すべての地域でCMC/医薬品の品質に関する書類が必要です。
- 臨床試験を開始し、実施する。
- 新規販売承認申請書(MAA、NDA、BLAなど)を提出する場合
- 製品のライフサイクル管理および承認後の変更に関する規制要件を満たすために
このモジュールには、原薬(DS)と製剤(DP)に関する書類一式が含まれています。
ドラッグ・サブスタンス(DS
医薬品のセクションは、物理的、化学的、または生物学的特性を含む記述、DS製造者の名前と住所、許容限界値、および医薬品の同一性、強度、品質、および純度を保証するために使用される分析方法によって提示される。
また、毒物学的試験および提案されている臨床試験中の原薬の安定性を裏付ける情報も含まれています。
ドラッグ・プロダクト(dp
医薬品の製造に使用されるすべての成分のリスト(不活性化合物の合理的な代替品を含む場合がある)で、医薬品に現れることが意図されている成分と、現れないかもしれないが製造工程で使用される成分の両方を含むものは、このセクションに記載されている。
以下は、薬事申請書類に記載する必要のある医薬品の主要情報です。
- 合理的なバリエーションを含む、(治験薬または市販薬)医薬品の定量的な組成。
- 医薬品メーカーの名前と住所。
- 製造およびパッケージング手順の説明
- 医薬品の同一性、強度、品質、および純度を保証するために使用される許容限界および分析方法。
CTD-Module 4
非臨床試験報告書
個々の動物のデータの適切な位置は、規制当局に提出される申請書の共通技術文書の試験報告書の中にある関連セクションです。モジュール4は、ICHの原則に基づいて米国およびEU向けに整合化されており、試験報告書に必要なすべてのセクションおよびサブセクションが含まれています。ICHガイドライン Common Technical Document (CTD)の参考文献。
CTD-Module 5
臨床試験報告書
Module 5 section this is the structure and content of clinical study reports.CTDのこの部分は、ヒト用医薬品の登録のための共通技術文書(CTD)内のヒト・臨床試験報告書、その他の臨床データ、および参考文献を提示したものである。これらの要素は、製造販売承認申請の準備と審査を容易にするものである。モジュール5は、ICHの原則に基づいて米国とEUで整合化されており、試験報告書に必要なすべてのセクションとサブセクションが含まれています。EUガイドラインの参照先 Common Technical Document (CTD).
規制当局への申請書類の作成にサポートが必要ですか?閲覧・相談 フリーランスの規制関連ライター をKolabtreeに掲載しました。
その他の参考資料
- https://www.ich.org/page/ctd
- https://www.fda.gov/media/128163/download
- https://www.fda.gov/media/135573/download
- https://ec.europa.eu/health/sites/default/files/files/eudralex/vol-2/b/update_200805/ctd_05-2008_en.pdf
- https://ec.europa.eu/health/sites/default/files/files/eudralex/vol-1/dir_2001_83_consol_2012/dir_2001_83_cons_2012_en.pdf
- https://eur-lex.europa.eu/legal-content/EN/TXT/PDF/?uri=CELEX:02001L0083-20121116&from=DE
- https://eur-lex.europa.eu/legal-content/EN/TXT/PDF/?uri=CELEX:02004R0726-20130605&from=EN
- https://ec.europa.eu/health/sites/default/files/files/eudralex/vol-1/reg_2000_141_cons-2009-07/reg_2000_141_cons-2009-07_en.pdf
- https://www.ema.europa.eu/en/human-regulatory/research-development/clinical-trials-human-medicines
- https://eur-lex.europa.eu/legal-content/EN/TXT/?uri=celex%3A32006R1901
- https://clinicaltrials.gov/ct2/home
Kolabtree helps businesses worldwide hire freelance scientists and industry experts on demand. Our freelancers have helped companies publish research papers, develop products, analyze data, and more. It only takes a minute to tell us what you need done and get quotes from experts for free.

Unlock Corporate Benefits
• Secure Payment Assistance
• Onboarding Support
• Dedicated Account Manager
Sign up with your professional email to avail special advances offered against purchase orders, seamless multi-channel payments, and extended support for agreements.


