



Shreya Chenni, フリーランスの規制関連ライター for medical devices, provides a 10-minute guide to FDA design controls for your 医療機器.
医療機器のリコールの主な原因の一つは、FDAが指摘した設計管理の不備です[3,3a]。その後、製造前の管理が機器GMP規制に追加されました。設計管理とは、設計・開発プロセスに組み込まれた、相互に関連する一連の実務と手順、すなわちチェックアンドバランスのシステムです。設計管理は、製造に移された設計が、意図された用途に適した機器になる可能性を高める。
適用性
すべてのクラスII、IIIおよび以下のクラスI機器は、デザインコントロールの対象となります。
- Devices automated with computer software
- 868.6810 気管気管支吸引用カテーテル
- 878.4460 手袋、外科医用
- 880.6760 拘束、保護
- 892.5650 システム、アプリケータ、放射性核種、手動
- 892.5740 線源、放射性核種による遠隔治療
- コンピュータソフトウェアで自動化されたデバイス
- 気管気管支吸引カテーテル
- サージングローブ
- 保護拘束
- システム、放射性核種、アプリケーター、マニュアル
- 線源、放射性核種による遠隔治療
Design controls apply to all D&D activities – for novel or improved devices being developed in the pre-market phase, as well as for changes to existing, marketed devices. Design controls do not apply to リサーチ activities conducted during the proof of concept stage.
Design controls can be applied to any 製品開発 process. The following flowcharts are examples of the design controls applied to a traditional Waterfall design process and V-model process for Software (SW).
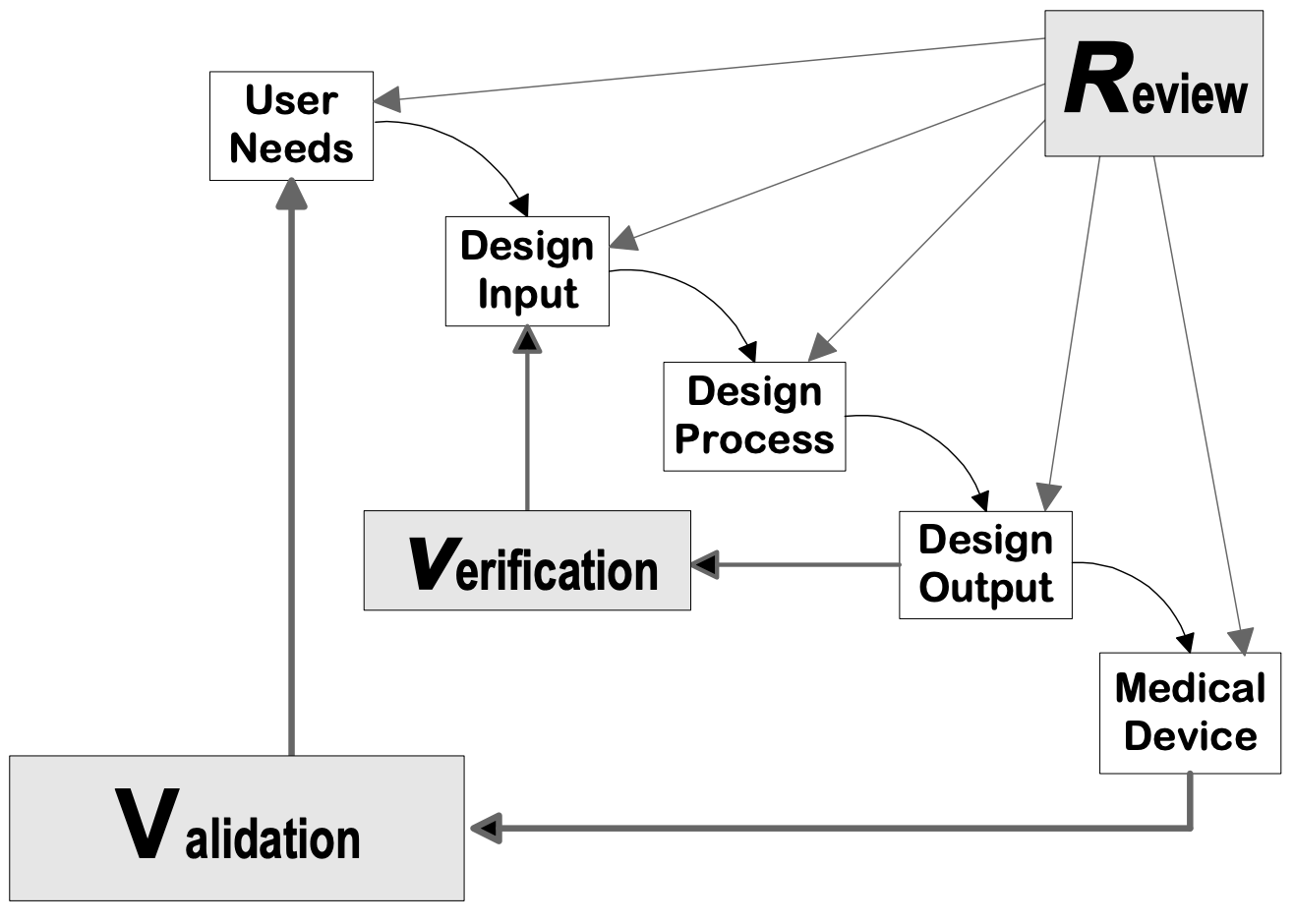
出典医療機器メーカー向けデザインコントロールガイダンス、CDRH-FDA
デバイス設計のフェーズ
D&D計画段階
D&D計画は確立され、維持されるべきである。この計画は、設計・開発活動を記述または参照し、実施のための責任を割り当てること。最低限、FDAは以下の内容を計画に盛り込むことを推奨している。
- D&Dプログラムの目標と目的
- デザイン活動の責任の明確化
- 主要なタスク、成果物を特定し、各タスクの責任者を割り当てる。
- 主な開発期間に合わせた主要タスクのスケジューリング
- 主要なレビューと決定ポイントの特定
- 査読者、査読チーム、査読者が従うべき手順の確認
- 設計書の管理
- 告知活動
この計画は、デザインや開発の進展に応じて見直し、更新し、承認されなければなりません。
設計入力段階
This is the starting point for product design. The medical device is designed and developed to meet the user requirements. Gather the user requirements from various sources such as customer surveys, feedback from the physicians, complaints. These requirements are transferred into design inputs.
- デザインの入力です。 これらは、デバイスの設計に使用される、デバイスの物理的および性能上の要求事項です。設計入力段階では、ユーザーの要求を製品の要求に変換します。設計入力を定義する際には、規制要件が考慮されます。設計入力要件は、包括的で、曖昧さがなく、客観的に検証可能でなければなりません。入力要件は、3つのカテゴリーにまとめることができます。
- 機能的要求事項とは、その機器が何をするのかを説明するものです。例えば、以下のようなものです。 ユーザーの指示により、車椅子が前方に移動すること
- 性能要件:その機器がどの程度、どのような性能を持つべきかを規定します。例えば、以下のようなものです。 車椅子は、前進方向に2m/sの速度で動くこと
- インターフェース要件:ユーザー/患者インターフェースなど、外部システムとの互換性に重要なデバイスの特性を指定します。例えば 車椅子には、方向を示すシンボルのボタンが付いていること。
- ここでは、ユーザーのニーズを定義し、それをデザインのインプットに変換した例を紹介します。
- ユーザーニーズ
デバイスはポータブルで、ブルートゥースに対応していること
- ユーザーニーズ
-
- デザイン入力
準拠すべき適用可能なFDA承認または国際規格を特定する。などです。
-
-
- IEEE ANSI C63.27-2017 無線の共存性の評価に関する米国国家規格
- AAMI TIR 69: Association for the Advancement of Medical Instrumentation – リスクマネジメント of Radio-frequency Wireless Coexistence for Medical Devices and Systems (2017)
- IEC 60601-1-2 Edition 3: 2007:医療用電気機器 - パート 1-2:安全性に関する一般要求事項 - 付帯規格電磁両立性-要求事項と試験
- UL 2054 - 家庭用および商業用バッテリーの規格
-
パフォーマンスやその他の具体的なインプットをリストアップします。などです。
-
-
- デバイスはDC電源であること
- Bluetoothモジュールを使用すること
- 重量:約6lbsまたは6lbs+/-2lbs(入力は検証を確実にするために定量的な制限を設けるべきである)
-
設計出力段階
デザインアウトプットとは、各設計段階における設計作業の結果、および設計作業全体の終了時に得られるものである。設計アウトプットの例としては、エンジニアリング図面、ラベリング、作業指示書、その他の製品仕様がある。その他の設計成果としては、リスク分析の結果、検証活動の結果、生体適合性試験の結果、ソフトウェアのソースコードなどがある。
設計出力は、担当者によるレビューと承認の前にリリースしてはいけません。また、デザインアウトプット/インプットの承認後にデバイスに変更を加える場合は、関係者によるレビューと承認を経て管理されることに留意する必要があります。 このフェーズの終わりには、デザインレビューが必要です。
デザインレビュー段階
デザインレビューは、設計出力フェーズの後に行われるべきである。デザインレビューは、確立された手順に従い、設計履歴ファイル(DHF)に文書化されるべきである。各デザインレビューの参加者は、すべての機能グループの代表者であるべきである。
正式なレビューは、プロジェクトの重要なマイルストーンの終わりに実施することが推奨されます。一般的に、デザインレビューは、デザインアウトプットフェーズ、V&Vフェーズ、デザイントランスファーフェーズの後に行われます。これは、デバイス開発の複雑さにもよります。FDAは少なくとも1回のデザインレビューを要求しています。
設計検証段階
設計検証とは、設計出力が設計入力を満たしていることを客観的な証拠によって確認することです。 基本的には「設計入力=設計出力」です。 検証活動は、確立された手順に従って実施されるべきである。検証の例としては、EMC・電気試験、目視検査、非臨床試験活動、プロセスや設計のフォールト ツリー解析、故障モード・影響解析などがあります。検証は、製品仕様の技術的要求事項が満たされていることを確認するものです。すべての検証活動は文書化されなければなりません。
トレーサビリティーマトリックス。 本ドキュメントは、デザインのインプットとアウトプットを表形式で記載しています。各入力に対して、対応する出力が参照される。この検証方法は、インプットとアウトプットが両方ともドキュメントである場合に使用されます。
デザインバリデーション段階
デザインバリデーションとは、仕様(規定された要求事項)がユーザーのニーズや意図された用途に適合していることを客観的な証拠によって証明することです。
- プロセスバリデーションとは、あるプロセスが所定の仕様を満たす結果や製品を一貫して生み出すことを、客観的な証拠によって証明すること。
- デザインバリデーションとは、デバイスの仕様がユーザーのニーズや意図した用途に適合していることを客観的な証拠によって証明することです。
バリデーションは通常 実際の環境下または模擬環境下で実施. Examples of validation include 臨床試験バリデーション活動の結果及び/又はバリデーション報告書は、設計履歴ファイル(DHF)の一部とし て文書化する。バリデーション活動の結果および/またはバリデーション報告書は、設計履歴ファイル(DHF)の一部となるよう文書化する必要があります。
設計移管フェーズ
V&Vステージの完了後、設計移管が行われます。これは、デバイスの品質を確保するために、デバイスの設計を製品仕様に移すことを含みます。この段階は非常に重要です。なぜなら、デバイスの生産が始まると、設計変更管理の対象となり、問題が発生すると金銭的な損失につながる可能性があるからです。設計移管は、確立された手順に従って行われなければなりません。また、設計移管を開始する前に、製品仕様を含む文書がレビューされ、承認されていることを確認する必要があります。
設計変更フェーズ
設計変更の管理は、設計移管時に始まり、ライフサイクルを通じて継続されます。設計移管後の設計変更は、確立された手順に従って実施されるエンジニアリング・チェンジ・ノーティス(ECN)の対象となります。設計変更の際には、リスクマネジメントレポート、使用説明書、検証・妥当性確認レポートなどの関連文書を再検討し、更新することが必要です。
デザインヒストリーファイル(DHF)
DHF は、米国 FDA に固有のものです。ISO 13486:2016 は、製造者が DHF を維持することを要求していない。
プロジェクトごとに、各フェーズの成果物をまとめた「デザインヒストリーファイル」を作成しています。また、最新の製品情報も含まれている。設計・開発文書は、必要に応じて容易に入手・アクセスできるものとする。設計・開発契約では、メーカーの設計情報に対する権利を明示し、設計文書の形式と内容に関する基準を定めるべきである。
実際には、デザインコントロールによって、マネージャーとデザイナーはデザインプロセスの可視性を向上させることができます。視認性の向上により,マネージャーは設計プロセスをより効果的に指揮することができるようになる.すなわち,問題を早期に認識し,修正を行い,リソースの割り当てを調整することができるようになる.設計者は,ユーザーや患者のニーズに対する設計の適合性についての理解を深めることができ,また,設計プロセスにおけるすべての参加者間のコミュニケーションや調整が改善されるという利点がある.
医療機器におけるFDAデザインコントロールの理解と導入にお困りですか?経験豊富な 医療機器コンサルタント をKolabtreeに掲載しました。
リファレンス
- FDA、医療機器メーカー向けデザインコントロールガイダンス
- メディカル・レギュラトリー・プラクティス、インターナショナル・パースペクティブ、Val Theisz
- デザインコントロール(Joseph Tartal氏によるプレゼンテーション
3a. フェデラルレジスター / Vol.61, No.195
Kolabtree helps businesses worldwide hire freelance scientists and industry experts on demand. Our freelancers have helped companies publish research papers, develop products, analyze data, and more. It only takes a minute to tell us what you need done and get quotes from experts for free.

Unlock Corporate Benefits
• Secure Payment Assistance
• Onboarding Support
• Dedicated Account Manager
Sign up with your professional email to avail special advances offered against purchase orders, seamless multi-channel payments, and extended support for agreements.


