



An exhaustive guide to regulatory compliance for IVD manufacturers, written by Sundeep Agarwal, experienced IVDR consultant.
What is the IVDR?
The European Commission’s (EC) In Vitro Diagnostic Regulation (EU IVDR 2017/746) is a ‘legislative framework’ and a way forward towards global IVD safety, which assures that only reliable and effective IVDs are in the market. The European Commission is trying its best to make the healthcare system safer and error free in terms of diagnosis or outcomes.
The in vitro diagnostic medical devices (IVDD), 98/79/EC was a directive while IVDR is a legislation (regulation) applicable to all Economic Operator (EO) i.e., manufacturers, importers, users, notified bodies and national authorities in the European Economic Area (EEA) and those non-EU-manufacturer and suppliers placing or planning to distribute IVD in the European market.
IVDR consist of 113 articles (10 chapters) and fifteen annexures in comparison to 24 articles, ten annexures of IVDD. No doubt, IVDR is a lengthy piece of regulation and considerably stringent but then the good part is that, it is more transparent with the regulatory changes and requirements.
It emphasizes on life cycle-based approach. It will be applied from 26 May 2022 and economic operators (including non-EU manufacturer) are expected to proactively prepare themselves towards planning and implementation of the same. Every stakeholder in the process shall now be equally responsible for the European Economic Area (EEA) in-vitro diagnostics market.
- The first and foremost thing an organization should do is to organize training program (online or onsite, as applicable) on EU IVDR so that everyone in the organization is aware of the necessary changes.
- An official communication should be followed by, to all the supplier, sub-contractors, or service provider about the process and their obligations.
- Perform a gap assessment to check the availability of their resources, a competent team to update the technical documentation required under EU IVDR. Being ISO 13485: 2016 certified would be an added advantage to establish compliance.
- It is advisable (If needed) to engage a subject matter expert or an external consultant from the very early stage of transition because ‘a stitch in time saves nine’.
- This blog will provide a detail outline and practical tips to comply with the expectation by the notified bodies and competent authorities as described in the various articles and annexures under EU IVDR 2017/746.
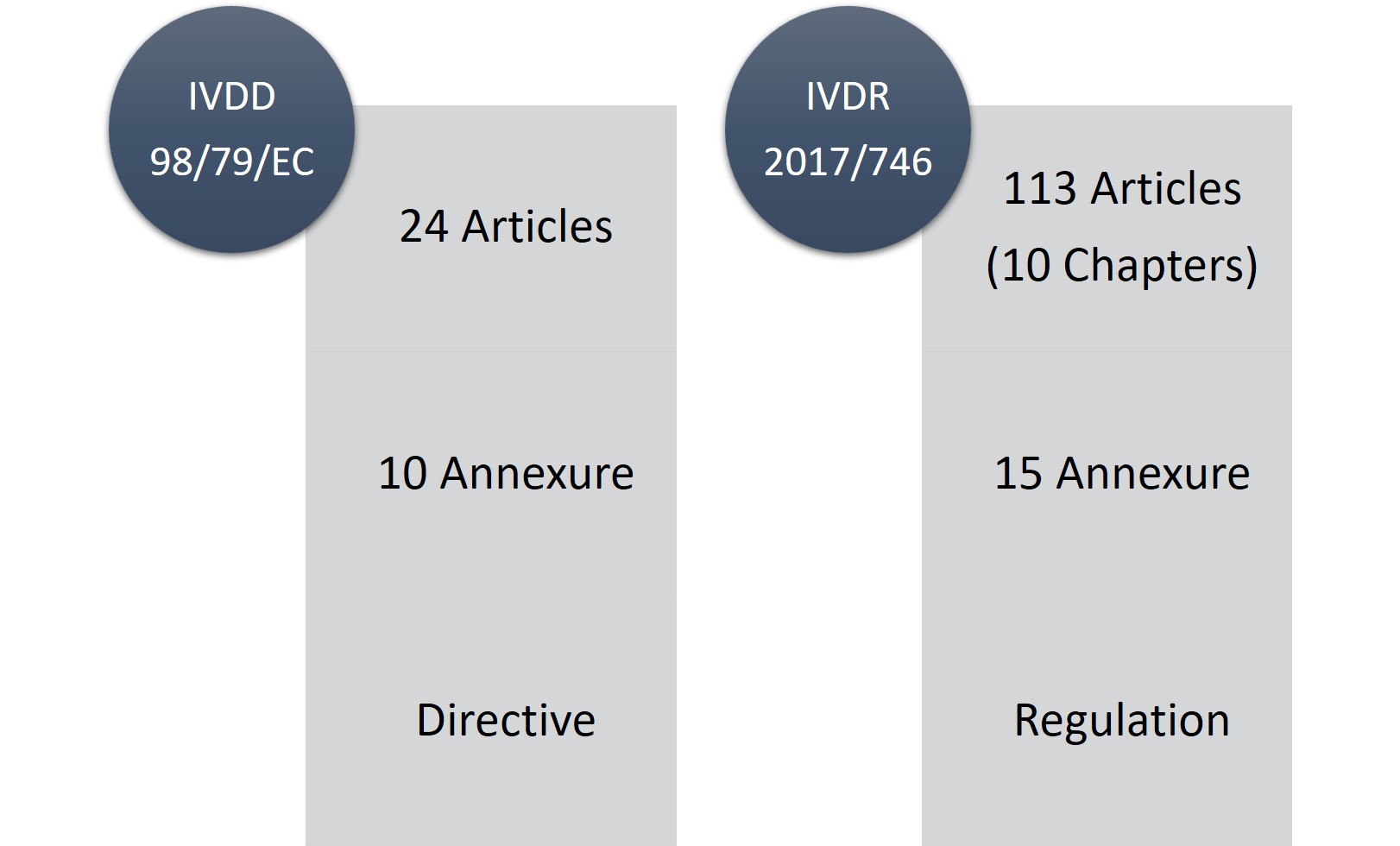
Figure1: IVDD vs. IVDR
1. Preparing for IVDR compliance and commercial changes
The foremost business decision for an organization would be to conclude if they want to continue to place their IVD in the European Economic Area (EEA). If the answer is ‘yes’, then one has should obtain estimates (cost), timelines, scope of audit, product code, etc. from a NB at the earliest possible. A shift from a directive to regulation calls for a mandatory compliance and robust technical documentation to establish safety and efficacy and to achieve CE Certification. IVDR largely relies a lot more on the clinical evidences i.e., scientific validity, analytical performance and clinical performance to establish the safety and efficacy.
The involvement of a notified body (NB) in the process of CE certification will be a prominent feature of the regulation. This also indicates an additional investment for the economic operator which may indirectly increase the cost of product.
An appointment of a “Person responsible for regulatory compliance (PRRC)” in accordance with Article 15 of EU IVDR 2017/746 is now mandatory; who shall assure the conformity of QMS, declaration of conformity, technical documentation, post market surveillance and reporting of adverse events are in compliance to EU IVDR.Manufacturers should ensure that the entire transition (including new certification application) is completed before the expiry of their existing IVDD Certificate or Self-certified Declaration of conformity. Certificates issued by notified bodies in accordance with IVDD 98/79/EC from 25 May 2017 shall become invalid after 27 May 2024. Be aware of the new timeline for application as per the EC official press release [1] dtd.20th December 2022.
2. Clear understanding of the classification
Reconsider the new classification rule under Annex VIII of IVDR and check if it has affected your previous classification.
Performing a correct classification is essential before preparing for the CE certification process. Unless we are able to do so, the conformity route will be unclear and delays or invalidate our efforts to comply with IVDR requirements.
IVDR is a risk-based approach to classify device with increased notified body and competent authority controls. The Regulation identifies four risk classes: Class A (lowest risk), Class B, Class C, and Class D (highest risk) while Annex VIII define seven classification rules to correctly classify the products. A unique feature of the IVDR is that, Software is also classified under Implementing rule 1.4 of Annex VIII, which states “Software, which drives a device or influences the use of a device, shall fall within the same class as the device. If the software is independent of any other device, it shall be classified in its own right[2]”. This indicates the scope for Software to be regulated under IVDR. And manufacturer has to also perform software verification and validation (Annex II, 6.4) accordingly.
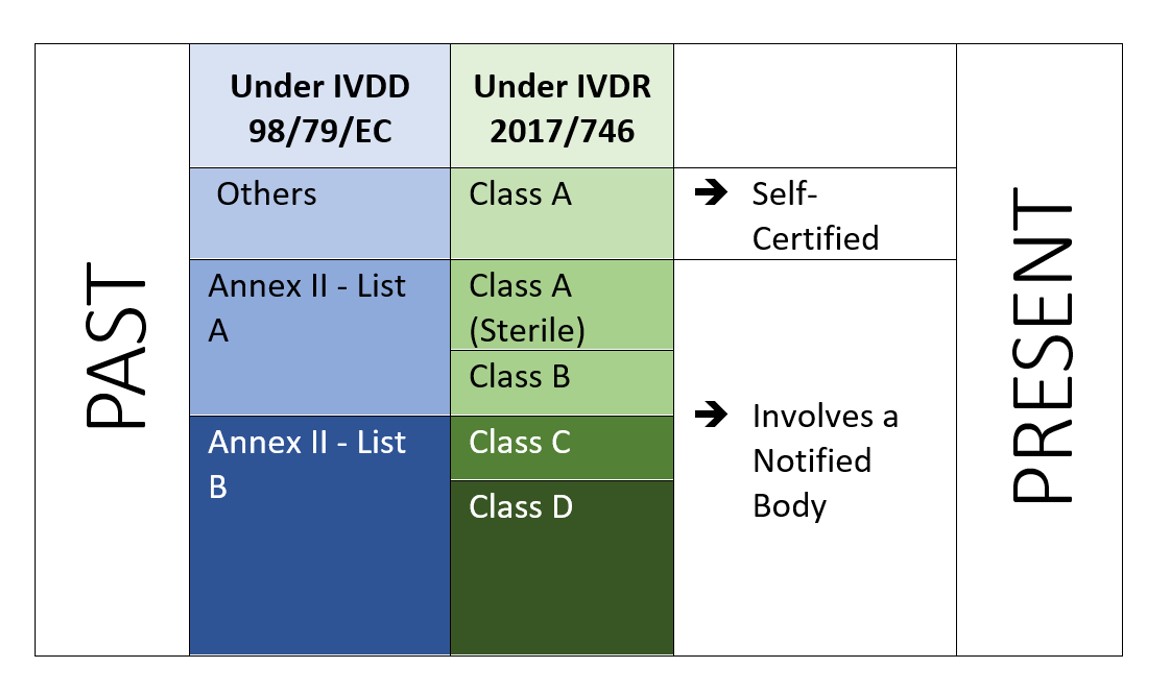
Figure 2: Risk Based Classification under IVDR 2107/746
3. Involvement of notified body
The role of a notified body (NB) would be one of the core elements and hence a larger number of manufacturers would now require to be audited and certified by a notified body vis-à-vis traditional method of “self-certification”.Economic Operators should carefully decide on the conformity assessment route (Annex IX, X, XI of EU IVDR[3]).
IVDR not only demands additional investment but has to assure that their technical documentation and quality management system meets the new requirements of IVDR. Under the IVDD, most of the IVDs are self-certified (92%), and do not require the involvement of a Notified Body (except 8% of the total IVD place in the market[4]). While under the new IVDR, the scenario is not the same.
According to a study “The impact of the new European IVD-classification rules on the notified body involvement” by National Institute for Public Health and the Environment, Bilthoven (Netherlands) RIVM Letter report 2018-0082, A. van Drongelen et al., nearly 85% of all IVDs will be requiring Notified Body involvement, leaving only 15% of IVDs eligible for self-certification[5].
This also means In Vitro Diagnostics (IVDs) manufacturer will now experience a major shift to comply with the new classification and certification process. Further depending on the intended use of the devices and risks class, manufacturer need to identify a designated NB which who might be able to audit them and get their products certified. Highest risk IVD (Class-D) would require EU reference lab, or expert panels to verify the performance claim additionally to the involvement of a Notified Body(NB) or Competent Authority (CA). Currently there only six notified bodies designated under EU IVDR. Do not wait to start your application process to avoid unexpected delays due to unavailability of a Notified Body.

Figure 3: List of designated notified body under IVDR[6]
4. Establishment of quality management system (QMS)
IVD manufacturers are expected to establish a robust and reliable quality management system (QMS) within their premises. It’s a general obligation of a manufacturer under Article 10 of IVDR. Quality management system is one essential requirement among various other, without which a manufacturer won’t be able to get approved.
QMS is to ensure that manufacturing, change control, customer complaints, resource management, supplier &sub-contractors’ controls and validation, performance evaluation, quality test, UDI Labelling, Post market surveillance etc. are according to approved QMS and Post Market Surveillance (PMS) plans.
The PRRC has to ensure the manufacturer has met the requirements of Article 10 to “self-certify” (issuance of Declaration of conformity in accordance with Annex IV) Class A IVD when a Notified Body (NB) is not required in the process.
5. Be prepared for disruption in the supply chain
Throughout the world, manufacturer depends largely on their supply chain and raw material to produce and deliver IVDs that are safe, accurate, and effective forthe intended use. Hence regulatory and quality concerns are also evolving to a higher level when it comes to the suppliers and sub-contractors’ controls. Manufacturer are therefore expected to proactively communicate the supply chain about their obligations and responsibilities of the suppliers and subcontractors. Legal manufacturer shall demonstrate adequate supplier control and monitoring, assure the supply chain is in compliance to the regulatory aspects of IVDR, reconsider the need for data integrity and quality of supplier data, implement robust supplier risk management and performance monitoring and periodically audit the supplier based on the associated risk to the finished products. The regulators and notified bodies are emphasizing on the legal manufacturers to clearly document the level of supplier controls and demonstrate with evidence that they have the potential to mitigate the risk of the product or service provided by the supplier.
6. Ensuring audit and inspection readiness
According to Article 88 of IVDR, Market Surveillance Activities, Competent authorities shall carry out both announced (unannounced) inspections on the premises of economic operators as well as suppliers and/or subcontractors and when necessary, at the facilities of professional users. While manufacturer shall include information on identification of all sites, including suppliers and sub-contractors, where manufacturing activities are performed in the Technical Documentation of Design & Manufacturing information. Notified bodies (NB) performing QMS audit shall identify links between and allocation of responsibilities among the various manufacturing sites, and their suppliers and/or subcontractors. This information will be considered while the NB specifically wants to audit any of those suppliers or subcontractors or both. The premises of the manufacturer’s suppliers, when considered to be significantly affecting the conformity of finished devices shall be essentially audited by the NB (in particular when the manufacturer cannot demonstrate sufficient control over its suppliers).
7. Plan to handle unannounced audits
Under post-certification monitoring, the NB shall proceed with unannounced on-site audits of manufacturers and their subcontractors or suppliers carrying out product tests and the monitoring of compliance with any conditions binding manufacturers and associated with certification decisions, such as updates to clinical data at defined intervals.Further, the notified body shall randomly perform at least once every five years unannounced audits on the site of the manufacturer and, where appropriate, the site of the manufacturer’s suppliers and/or subcontractors, which may be combined with the periodic surveillance assessment.
8. Strengthen post-market surveillance activities
Manufacturers are strongly recommended to strengthen their post-market surveillance requirements and develop mechanism to coordinate between EU member states on vigilance and market surveillance. Under surveillance assessment applicable to class C and class D devices (Annex IX), the notified body shall periodically, at least once every 12 months, carry out appropriate audits and assessments. It shall include audits on the premises of the manufacturer and suppliers and/or subcontractors as applicable. Manufacturer shall essentially develop a procedure for recording and reporting of incidents and Field Safety Corrective Action (FSCA).
9. Unique device identifier (UDI) & EUDAMED
Manufacturer shall have to establish a system for UDI to identify and facilitate traceability of devices. ‘Device Identifier’ and ‘Production identifier’ shall be on the labels to enhance traceability in EU market.One may refer to a list of accredited issuing Entity (IE) such as GS1, HIBCC, ICCBBA, IFA GmbH to operate a system for the assignment of UDIs. Currently the mentioned IEs are valid from 27th June 2019, but it will be wise to confirm their validity while making a final decision on implementing them.
European Database on Medical Devices (EUDAMED) will provide an overview of all medical devices available in the European Union. It consist of six modules related to:
- Actor registration,
- Unique device identification (UDI) and device registration,
- Notified bodies and certificates,
- Clinical investigations and performance studies,
- Vigilance and Post market surveillance, and
- Market surveillance.
To ensure an improved transparency through a comprehensive EUDAMED, parts of Economic Operators information will be publicly accessible. While confidential information will be accessible only to Economic Operator, Sponsors, Notified and Competent Authorities of EU member states.
10. Requirements for “in-house devices”
Health institution developing ‘in-house devices’ (or ‘laboratory-developed tests’) which are meant to be used by the same health institution shall not be marketed or sold to other legal entity. Such devices may be used for the diagnosis and treatment, especially for rare diseases. The institution is expected to comply with only the requirement of Annex I of IVDR (general safety and performance requirements), and exempted from rest of the regulation until 26 May 2024; provided the health institution meets a number of conditions set out in Article 5(5) of the Regulation and has an appropriate quality management system, which complies to the international standard setting out the quality and competence requirements for medical laboratories (EN ISO 15189) or other national provisions, and is able to justify that target patient group’s specific needs cannot appropriately be met by an equivalent device available on the market.
References
[1] EC official press release dtd. 20th Dec 2021, Progressive roll-out of the In Vitro Diagnostic Medical Devices Regulation. Can be accessed at https://ec.europa.eu/commission/presscorner/detail/en/IP_21_6965 [2]REGULATION (EU) 2017/746 OF THE EUROPEAN PARLIAMENT AND OF THE COUNCIL of 5 April 2017 on in vitro diagnostic medical devices and repealing Directive 98/79/EC and Commission Decision 2010/227/EU; ANNEX VIII CLASSIFICATION RULES, 1. IMPLEMENTING RULES Point 1.4 Page 304 [3]Regulation (EU) 2017/746 of the European parliament and of the council of 5 April 2017 on in vitro diagnostic medical devices and repealing Directive 98/79/EC and Commission Decision 2010/227/EUANNEX IX Conformity Assessment Based On A Quality Management System And On Assessment Of Technical Documentation, Page 306, ANNEX X Conformity Assessment Based On Type-Examination, Page 314, ANNEX XI Conformity Assessment Based On Production Quality Assurance, Page 317
[4] Press release dtd. 14 October 2021, Brussels; Public health: Commission proposes a progressive roll-out of the new In Vitro Diagnostic Medical Devices Regulation [5]The impact of the new European IVD-classification rules on the notified body involvement; : a study on the IVDs registered in the Netherlands; van Drongelen A, de Bruijn A, Pennings J, van der Maaden T 32 p in English 2018, RIVM letter report 2018-0082 [6] The above list is based on data accessed dtd. 5th March 2021, for the latest updates on the list, you may access the official website of EC at https://ec.europa.eu/growth/tools-databases/nando/index.cfm?fuseaction=directive.notifiedbody&dir_id=35Kolabtree helps businesses worldwide hire freelance scientists and industry experts on demand. Our freelancers have helped companies publish research papers, develop products, analyze data, and more. It only takes a minute to tell us what you need done and get quotes from experts for free.

Unlock Corporate Benefits
• Secure Payment Assistance
• Onboarding Support
• Dedicated Account Manager
Sign up with your professional email to avail special advances offered against purchase orders, seamless multi-channel payments, and extended support for agreements.


