



Biologo computazionale e Kolabtree freelance Shaurya Jauhari writes about current challenges involved in conducting pathway analysis in bioinformatics and possible solutions to the problem.
In the contrasting era of “Imprecise Medicine” (propelling us to tune towards Precision Medicine) and inundation of biomedical data brought forth by advancements in instrumentation technologies, a lacuna persists that is largely premised over mapping data to information. The clinical experiments engender biomarcatori (technically list of genes or genomic regions more contemporarily) that have to be expounded for their biological implications. The current suite of tools that facilitate such an endeavor is less purposeful as it neglects the ipso facto, le interazioni spaziali del genoma dato il suo profilo di sistemazione all'interno del nucleo di ogni cellula eucariotica. Questo commento intende evidenziare la natura del problema, far luce sull'organizzazione del genoma, riflettere brevemente sugli attuali strumenti di mappatura e ipotizzare le possibili soluzioni.
Il diavolo è nei dettagli
Una serie di sforzi per affinare la risoluzione dei dati genomici sta spazzolando sotto uno schema cruciale di informazioni. Stiamo impellendo con ansia la verosimiglianza di un genoma $1000, anche se si preoccupa meno dell'analisi $100.000. Esiste un grande compendio di repertori che contengono le annotazioni dei risultati sperimentali e dei casi sotto un tipico studio biologico. Ci potrebbero essere definizioni che dichiarano le implicazioni biologiche di un gene, o il percorso di cui questi geni fanno parte, associato a una malattia. Di nuovo, questo tipo di archivi dinamici e informativi sono stati curati manualmente (in passato) e la gestione della conoscenza è stata rilevata da pipeline automatizzate che impiegano computer e ICT in generale. Questi database sono aggiornati con una saggezza scientifica orientata al consenso e hanno abbracciato una manciata di revisioni dall'inizio. Il condotto di mappatura dei risultati sperimentali alle loro implicazioni biologiche è fortemente ridotto, in gran parte perché la "vera" biologia sottostante viene ignorata.
Il nostro genoma, lungo in media circa 2 metri, è alloggiato nel nucleo di ogni singola cellula del nostro corpo. A causa della struttura ridotta di una cellula e soprattutto del suo nucleo, il genoma è impacchettato in modo un po' teso e molle. Questo permette che regioni del genoma, che sono piuttosto distanti da una prospettiva lineare, si avvicinino e interagiscano. Questo adagio è grossolanamente rifiutato dall'attuale suite di strumenti di arricchimento (mappatura) e quindi i risultati generati sono sproporzionati.
Le regioni del genoma fanno parte di "gruppi d'azione" più grandi o percorsi that are technically series of chemical reactions accounting for a phenotype; healthy or diseased. When a diseased state is examined, the investigators are on the lookout for the biomarkers that have potentially gone awry and have apparently transformed the organismal body from tonico a si è mosso. Immaginate di inseguire una malattia combattuta con informazioni non allineate.
 Dichiarazione del problema - Flusso di lavoro di una tipica analisi di arricchimento. C'è un certo
Dichiarazione del problema - Flusso di lavoro di una tipica analisi di arricchimento. C'è un certo
"idiomatismo" associato alla mappatura delle sequenze del genoma ai geni, e che orchestra il
risultati a valle.
Organizzazione del genoma
Come accennato prima, il genoma è abbastanza lungo da essere immagazzinato linearmente all'interno del nucleo di una cellula, per ogni cellula del nostro corpo o di qualsiasi altro organismo vivente. Piuttosto questa entità di 2 metri è schiacciata e stipata in una struttura apparentemente disordinata, con anelli, curve e vortici come si può immaginare. Questo senza fine stringa di nucleotidi o coppie di basi - Adenina (A), Citosina (C), Timina (T), e Guanina (G) strutturano topologie distinte all'interno del nucleo mentre si adattano all'esiguità. Formano anelli di cromatina, compartimenti/sottocompartimenti, domini/sottodomini che servono a uno scopo, secondo il tipo di cellula. (Si noti che i diversi tipi di cellule funzionano in modo diverso; una cellula nervosa ha altri affari da soddisfare rispetto a una cellula muscolare; ognuna ha un ruolo esclusivo da svolgere).
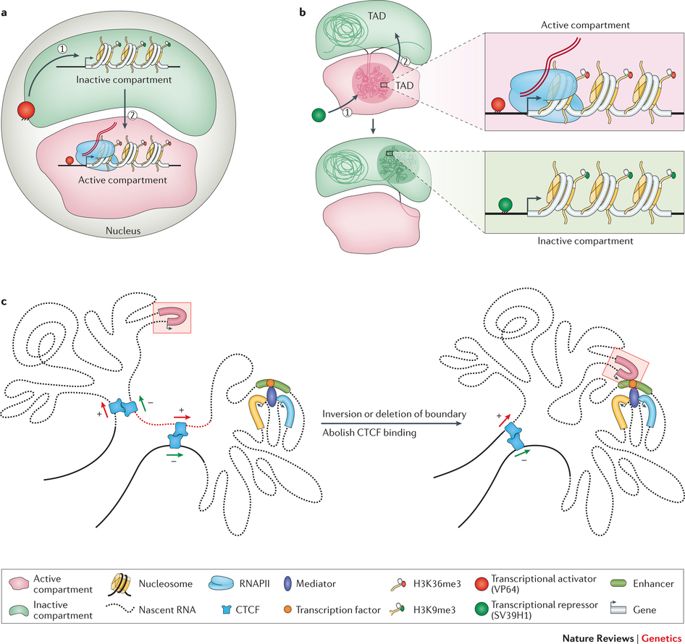
Il genoma si avvolge e si espande negli spazi oggettivi. (Credito: https://doi.org/10.1038/nrg.
2016.112)
Basi di dati del percorso
Esistono ontologie e banche dati variegate, tra cui la Kyoto Encyclopedia of Genes and Genomes (KEGG) (https://www.genome.jp/kegg/) e Gene Ontology (http://geneontology.org/I pochi strumenti di cui parlerò nella prossima sezione (e che sono spesso opinionati) generano termini di arricchimento che provengono "selettivamente" dai suddetti database. Sulla base dei loro valori di significatività statistica, si ricava se rappresentano veramente un fenotipo elencato o sono solo una questione di sviluppo casuale.(P.S. C'è uno scritto sui valori p che presumibilmente aiuterà i profani a capire l'idea di significatività statistica. Per favore segui il link https://sway.office.com/WkyHrPnVB8Ec3zPD?play a vostro piacimento).
Strumenti di arricchimento
L'analisi di arricchimento è un protocollo computazionale di novo regioni genomiche alle loro definizioni registrate nei database a cui si è accennato. Parlando degli strumenti (che fungono da tramite), essi sono classicamente strutturati sotto diverse teste, vale a dire analisi di sovrarappresentazione (ORA), punteggio di classe funzionale (FCS), metodi basati sulla topologia del percorso (PT) e metodi di interazione di rete (NI).
Analisi della sovra rappresentazione
L'analisi di sovrarappresentazione, attraverso il dogma della distribuzione ipergeometrica, valuta l'insieme dei geni differenzialmente espressi per quelli che potrebbero far parte di un percorso biologico. Fondamentalmente, un test ipergeometrico considera quattro attributi per raggiungere una decisione, vale a dire
- Numero totale di geni nel test considerato,
- I geni differenzialmente espressi,
- Geni nel percorso di destinazione sul numero totale di geni, e
- Geni differenzialmente espressi che si verificano nel percorso di destinazione.
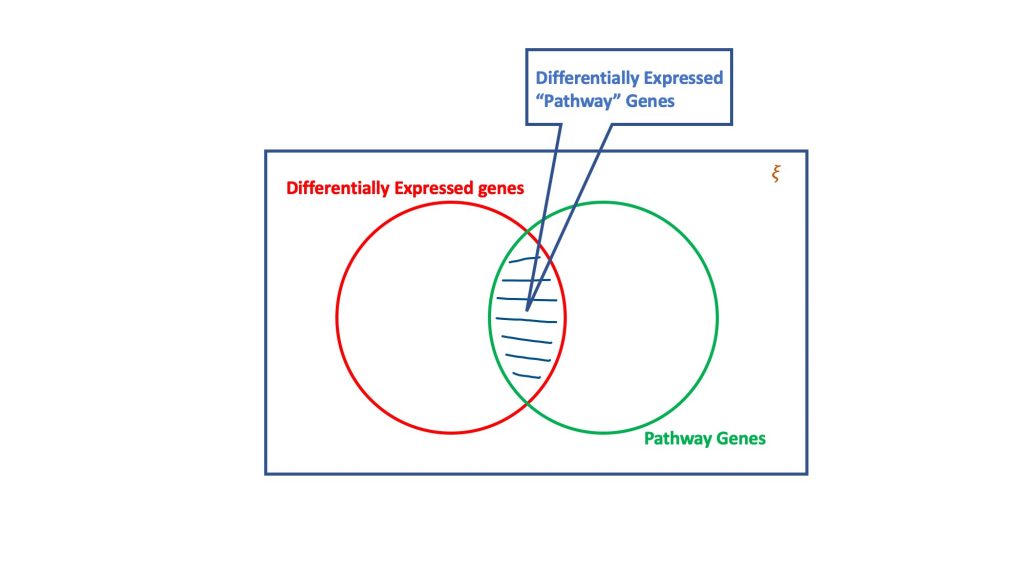
L'essenza del test ipergeometrico
Despite being a simple and straightforward methodology, ORA presents its own limitations.
- Democrazia in gioco; tutti i geni sono considerati allo stesso modoPerché questo è un problema? Supponiamo che i geni siano filtrati sulla base di fold-change. Setacciamo i geni che hanno abbracciato la differenza di espressione maggiore o uguale a 2 volte (pieghe), sia in direzione negativa che positiva. Anche se il minimo era 2 volte, questo flusso di lavoro avrebbe anche catturato i geni con cambiamenti di espressione erano 3 volte, 4 volte e oltre. Sicuramente, un gene con una disparità di espressione di 4 volte è più prudente di un gene con un cambiamento di 2 volte. Questa manifestazione è scontata da ORA.
- Considera solo i geni più significativiDi nuovo, consideriamo un gene con fold-change 1,9999 o p-value <0,0051113; comunemente, un p-value <0,05 è considerato statisticamente significativo. La metodologia ORA sorvola questo gene nel risultato finale. Chiaramente, c'è una perdita di informazioni e una mancanza di flessibilità. (P.S. Breitling et al. hanno affrontato questa situazione proponendo un'estemporizzazione per evitare le soglie. La revisione impiega un approccio iterativo che aggiunge un gene alla volta per compilare un insieme di geni per i quali un percorso è ottimamente significativo).
- Nessun gene funziona in modo isolatoCiò deriva dalle limitazioni sopra menzionate che trattando il gene come un'entità indipendente si perde il nocciolo del contributo poligenico verso un fenotipo. Un obiettivo dell'analisi dell'espressione genica potrebbe essere quello di chiarire le coorti di geni i cui modelli di espressione sono congruenti. Questa sinfonia evidenzia i geni funzionalmente affini o i geni che lavorano verso uno stato biologico comune.
- Percorsi reciprocamente indipendentiL'ORA presuppone anche che i percorsi non lavorino in tandem (o in successione). Questo è principalmente errato in quanto le serie di reazioni chimiche potrebbero benissimo precedere o procedere l'una con l'altra.
Punteggio della classe funzionale
Contrary to ORA, FCS methods subsume all the contextual genes as well as their association statistiche (fold-change, p-value) and compute a che corre punteggio di arricchimento per raggruppamenti di geni (basato su alcune conoscenze funzionali come Gene Ontology o KEGG pathways). es. GSEA dal Broad Institute (http://software.broadinstitute.org/gsea/index.jsp). Una tipica esecuzione FCS analizzerebbe il cambiamento di espressione dei geni complessivi nella lista (non la classifica per significatività statistica o altro) dei geni differenzialmente espressi in un esperimento. Il risultato principale dell'analisi di arricchimento del set di geni è un punteggio di arricchimento (ES) che riflette il grado in cui un set di geni è sovrarappresentato in cima o in fondo a una lista classificata di geni; perché in cima e in fondo? perché ci sono i geni più lontani dal normale, in termini di cambiamento di espressione. Un punteggio ES positivo per un set di geni (o un percorso target, GS) sarà indicativo dei geni della lista (GL) che cadono in alto (più upregolati; 1,2,3 ...), mentre un punteggio ES negativo significa che i geni componenti si trovano in basso (più downregolati; n-3, n-2, n-1, n, dove n è il numero totale di geni). P.S. L'ES diventa ES normalizzato (NES) quando si corregge il problema dei test multipli (false discovery rate, es. Metodo Bonferroni).
In sintesi, i metodi FCS sono notevolmente migliori dei metodi ORA,
- evitando il requisito di una soglia arbitraria per classificare i geni come significativi o non significativi.
- apprezzando le informazioni sull'espressione genica per seguire i cambiamenti sistematici nel percorso; questo rende conto dell'interdipendenza dei geni.
Tuttavia, anche i metodi FCS hanno alcune carenze.
- Poiché i percorsi sono analizzati in modo indipendente, i geni che regolano diversi percorsi potrebbero non essere contati.
- Molti metodi FCS classificano i geni in una lista sulla base dei cambiamenti nell'espressione genica. Uno scenario in cui la differenza tra i ranghi riflette una varianza disuguale (e possibilmente esponenziale) nell'espressione potrebbe forse essere una misura ingiusta.
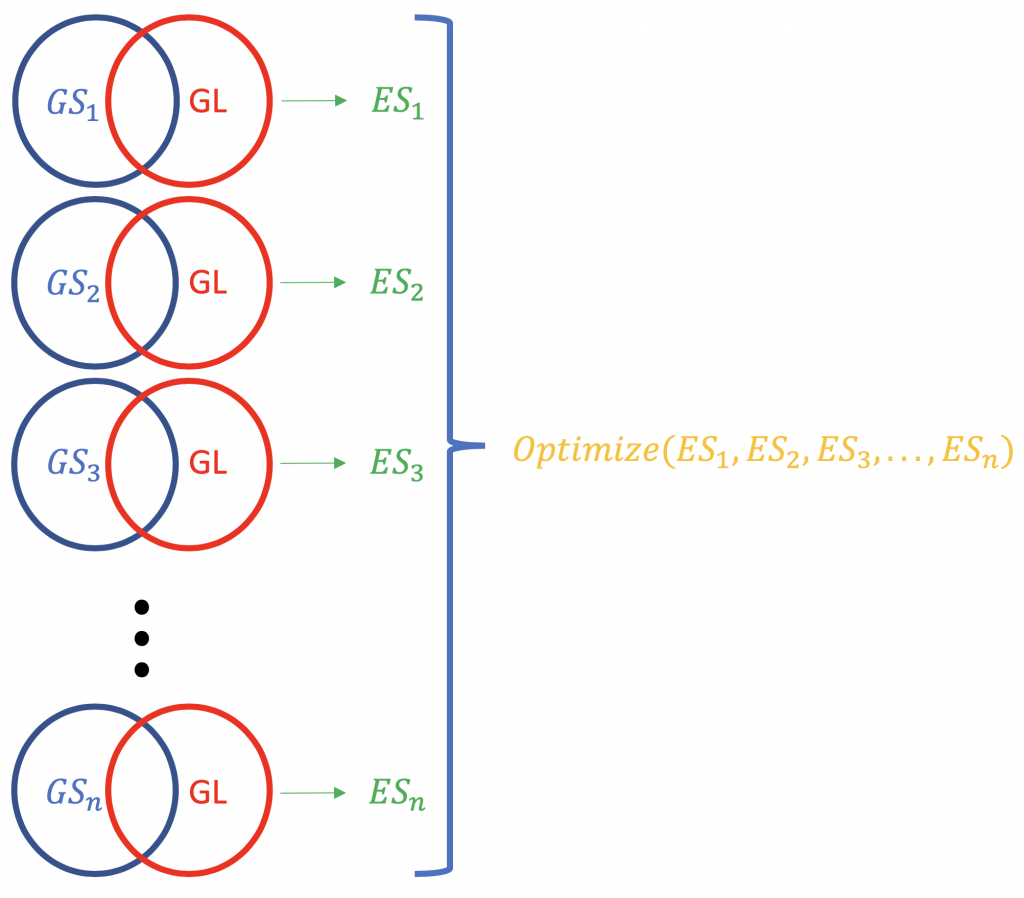
La normalizzazione dei punteggi ES relativi a una lista di geni.
Approcci basati sulla topologia del percorso
Una privazione cruciale dei metodi ORA e FCS è che essi ignorano la struttura dei percorsi. L'ordine dei geni che sono regolati in un percorso è essenziale per tracciare gli effetti causali. Comprensibilmente, ci potrebbero essere esattamente due percorsi con gli stessi componenti genici, ma la gerarchia di attivazione potrebbe essere completamente diversa. Se fosse stato per i metodi ORA/FCS, avrebbero portato a termini di arricchimento simili. Questo è un problema. I metodi Pathway Topology(PT) presuppongono l'esclusività della funzione a seconda delle interazioni specifiche, il che è anche conforme alla logica generale. Esempi di strumenti sono SPIA (https://www.bioconductor.org/packages/release/bioc/manuals/SPIA/man/SPIA.pdf), GGEA e PARADIGM. In generale, gli strumenti di questa categoria hanno un punteggio locale e uno globale. Il punteggio locale a livello di gene calibra i cambiamenti di fold nell'espressione del gene e dei geni a monte, mentre il punteggio globale misura la misura a livello di pathway per la parentela con il set di geni. Tuttavia, questo rende anche che i metodi PT sovradimensionano i dati per una particolare condizione/tipo di cellula.
Analisi basata sull'interazione di rete
Questa è una categoria piuttosto sottovalutata che è ancora poco implementata, nonostante la sua formulazione datata. Metodi come EnrichNet , NetPEA (http://www.dx.doi.org/10.1109/BIBM.2013.6732493) have been proposed close to a decade ago, but they haven’t gained much traction because of limited tools available. This facade constraints the dexterity of the theme as no improvements are documented. That turns out to be an open-ended ricerca problem.
L'apparente Panacea
Probabilmente ora avete un'idea dell'arricchimento fondamentale/analisi dei percorsi e del tipo di strumenti che la aiutano. Tuttavia, come ho detto prima, tutti gli strumenti disponibili (che rientrano nelle categorie contrassegnate) si basano su un parametro skewed di una finestra lineare attraverso la regione interrogata. Se i segmenti del genoma che intervengono rientrano in questa cornice, sono elencati come arricchitoaltrimenti no. Il vero affare è avere uno strumento che, forse, immette un centro e diametro di un ipotetico cerchio, come per evidenziare l'organizzazione 3D basata sulle interazioni del genoma per la regione.

GREAT offre diverse opzioni avanzate per specificare la dimensione lineare intorno al Transcription Start Site di un gene.
 Enrichr presenta una selezione diretta del tipo di genoma e del numero di geni nella regione lineare presunta.
Enrichr presenta una selezione diretta del tipo di genoma e del numero di geni nella regione lineare presunta.
Come si deduce dalla Figura 4, non c'è apprezzamento per la "vera" organizzazione spaziale del genoma. Questo è un problema e una profonda linea di faglia che persiste nell'attuale dominio dell'analisi di arricchimento. Nonostante ciò, c'è anche una forte rilevanza di fabbriche di trascrizioneidentificati come siti nello spazio nucleare che attirano elementi regolatori lontani per "fare festa in casa". Scherzando, osservo spesso che, come direbbe qualcuno che è arrabbiato con qualcuno o qualcosa, le fabbriche di trascrizione (personificate) potrebbero rimproverare il genoma".La trascrizione avverrà sul mio cadavere, nient'altro!". Il tema adiacente delle fabbriche di trascrizione è oggetto di qualche discussione futura adiacente. Tuttavia, materializza il dogma delle interazioni cis-regolatorie che è sbagliato nella pratica contemporanea.
Per concludere, vorrei sottolineare che l'analisi dei percorsi è una frazione cruciale, e spesso trascurata, della Bioinformatica tubazione. C'è sempre un margine per scalare le metodologie esistenti in tandem con i dati genomici, che si stanno evolvendo mentre parliamo. Quando il volume escalation di dati diventa disponibile per noi, non sarà solo un problema infrastrutturale, ma anche un blocco algoritmico.
Necessità di assumere un consulente bioinformatico? Lavora con gli scienziati freelance su Kolabtree. È gratuito pubblicare il tuo progetto e ricevere preventivi da esperti.
Esperti correlati:
Bioinformatica freelance | Genetica vegetale | Biologia dello sviluppo | Terapia genica | Cellule staminali |
Analisi dei dati di sequenziamento del DNA |Genetica animale | Interazioni tra farmaci | Genetica e genomica
Kolabtree helps businesses worldwide hire freelance scientists and industry experts on demand. Our freelancers have helped companies publish research papers, develop products, analyze data, and more. It only takes a minute to tell us what you need done and get quotes from experts for free.

Unlock Corporate Benefits
• Secure Payment Assistance
• Onboarding Support
• Dedicated Account Manager
Sign up with your professional email to avail special advances offered against purchase orders, seamless multi-channel payments, and extended support for agreements.


