



An exhaustive guide to conformità normativa for IVD manufacturers, written by Sundeep Agarwal, con esperienza Consulente IVDR.
Cos'è l'IVDR?
The European Commission’s (EC) In Vitro Diagnostic Regulation (EU IVDR 2017/746) is a ‘legislative framework’ and a way forward towards global IVD safety, which assures that only reliable and effective IVDs are in the market. The European Commission is trying its best to make the assistenza sanitaria system safer and error free in terms of diagnosis or outcomes.
I dispositivi medici diagnostici in vitro (IVDD), 98/79/CE era una direttiva mentre IVDR è una legislazione (regolamento) applicabile a tutti gli operatori economici (EO), cioè produttori, importatori, utenti, organismi notificati e autorità nazionali nello Spazio economico europeo (SEE) e quei produttori e fornitori non UE che collocano o progettano di distribuire IVD nel mercato europeo.
IVDR consiste di 113 articoli (10 capitoli) e quindici allegati in confronto a 24 articoli, dieci allegati di IVDD. Senza dubbio, l'IVDR è un regolamento lungo e considerevolmente rigoroso, ma la parte positiva è che è più trasparente con i cambiamenti e i requisiti normativi.
Enfatizza l'approccio basato sul ciclo di vita. Sarà applicato dal 26 maggio 2022 e gli operatori economici (compresi i produttori non UE) sono tenuti a prepararsi proattivamente alla pianificazione e all'attuazione dello stesso. Ogni attore del processo sarà ora ugualmente responsabile del mercato della diagnostica in vitro dello Spazio economico europeo (SEE).
- La prima e principale cosa che un'organizzazione dovrebbe fare è organizzare un programma di formazione (online o in loco, a seconda dei casi) sull'EU IVDR in modo che tutti nell'organizzazione siano consapevoli dei cambiamenti necessari.
- Una comunicazione ufficiale dovrebbe essere seguita da, a tutti i fornitori, subappaltatori, o fornitori di servizi circa il processo e i loro obblighi.
- Eseguire una valutazione delle lacune per verificare la disponibilità delle loro risorse, un team competente per aggiornare la documentazione tecnica richiesta dall'UE IVDR. Essere certificati ISO 13485: 2016 sarebbe un ulteriore vantaggio per stabilire la conformità.
- È consigliabile (se necessario) coinvolgere un esperto in materia o un consulente esterno fin dalle prime fasi della transizione, perché "un punto in tempo ne salva nove".
- Questo blog fornirà uno schema dettagliato e consigli pratici per conformarsi alle aspettative degli organismi notificati e delle autorità competenti come descritto nei vari articoli e allegati sotto l'UE IVDR 2017/746.
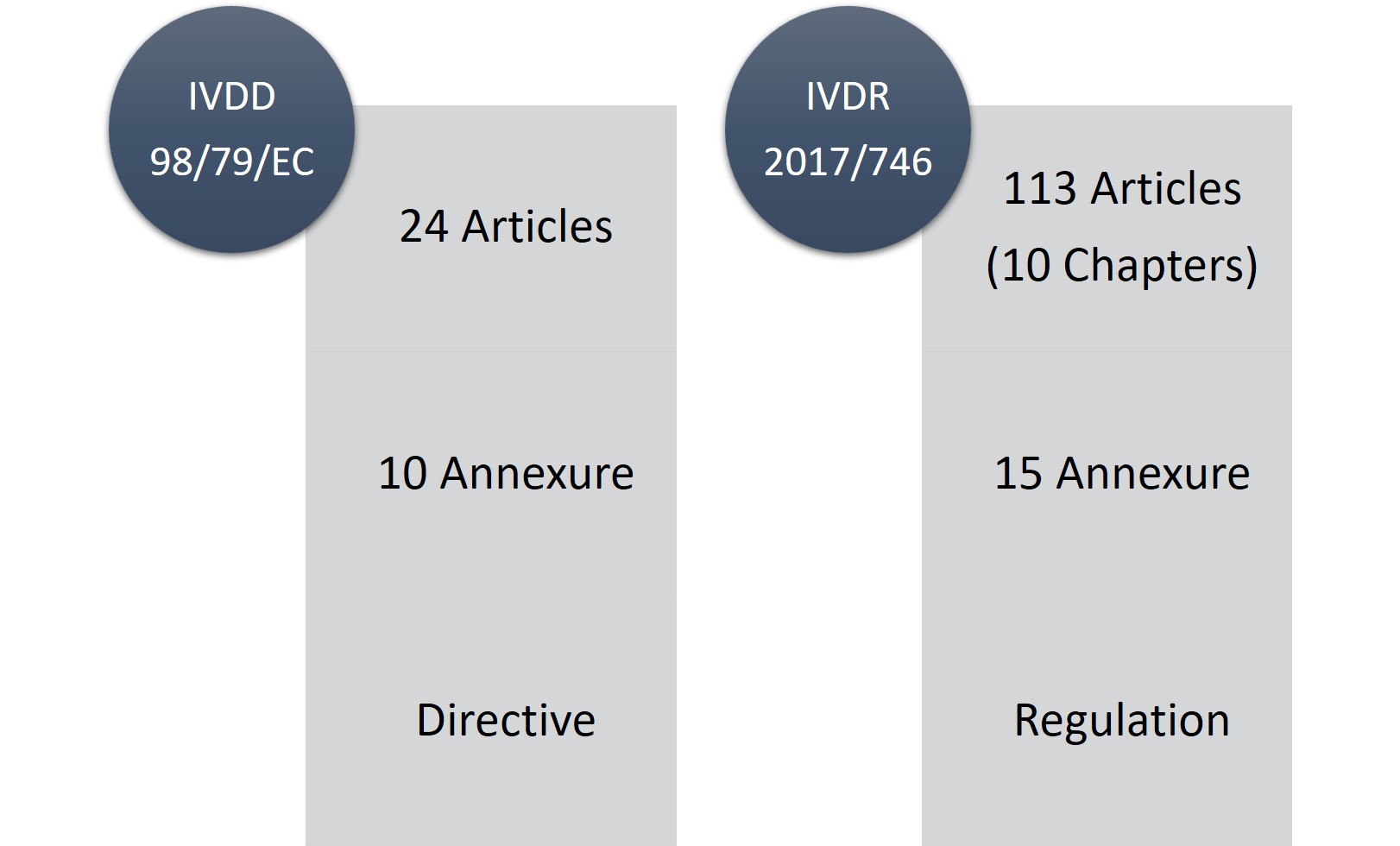
Figura1: IVDD vs. IVDR
1. Prepararsi alla conformità IVDR e ai cambiamenti commerciali
La principale decisione commerciale per un'organizzazione sarebbe quella di concludere se vogliono continuare a piazzare i loro IVD nello Spazio economico europeo (SEE). Se la risposta è "sì", allora si deve ottenere al più presto da un NB stime (costi), tempistiche, portata della revisione, codice prodotto, ecc. Il passaggio da una direttiva a un regolamento richiede una conformità obbligatoria e una documentazione tecnica solida per stabilire la sicurezza e l'efficacia e per ottenere la certificazione CE. IVDR si basa molto di più sulle evidenze cliniche vale a dire, validità scientifica, performance analitica e performance clinica per stabilire la sicurezza e l'efficacia.
Il coinvolgimento di un organismo notificato (NB) nel processo di certificazione CE sarà una caratteristica importante del regolamento. Questo indica anche un investimento aggiuntivo per l'operatore economico che può indirettamente aumentare il costo del prodotto.
Una nomina di un "Persona responsabile della conformità normativa (PRRC)" in accordance with Article 15 of EU IVDR 2017/746 is now mandatory; who shall assure the conformity of QMS, declaration of conformity, technical documentation, post market surveillance and reporting of adverse events are in compliance to EU IVDR.Manufacturers should ensure that the entire transition (including new certification application) is completed before the expiry of their existing IVDD Certificate or Self-certified Declaration of conformity. Certificates issued by notified bodies in accordance with IVDD 98/79/EC from 25 May 2017 shall become invalid after 27 May 2024. Be aware of the new timeline for application as per the EC official press release [1] dtd.20il Dicembre 2022.
2. Chiara comprensione della classificazione
Riconsideri la nuova regola di classificazione secondo l'allegato VIII dell'IVDR e verifichi se ha influenzato la sua precedente classificazione.
Eseguire una corretta classificazione è essenziale prima di preparare il processo di certificazione CE. Se non siamo in grado di farlo, il percorso di conformità sarà poco chiaro e ritarderà o invaliderà i nostri sforzi per rispettare i requisiti IVDR.
IVDR è un approccio basato sul rischio per classificare il dispositivo con maggiori controlli dell'organismo notificato e dell'autorità competente. Il regolamento identifica quattro classi di rischio: Classe A (rischio più basso), classe B, classe C e classe D (rischio più elevato) mentre l'allegato VIII definisce sette regole di classificazione per classificare correttamente i prodotti. Una caratteristica unica dell'IVDR è che il software è anche classificato in base alla regola di attuazione 1.4 dell'allegato VIII, che afferma: "Il software, che guida un dispositivo o influenza l'uso di un dispositivo, deve rientrare nella stessa classe del dispositivo. Se il software è indipendente da qualsiasi altro dispositivo, deve essere classificato a pieno titolo[2]". Questo indica l'ambito del software da regolare sotto IVDR. E il produttore deve anche eseguire la verifica e la convalida del software (allegato II, 6.4) di conseguenza.
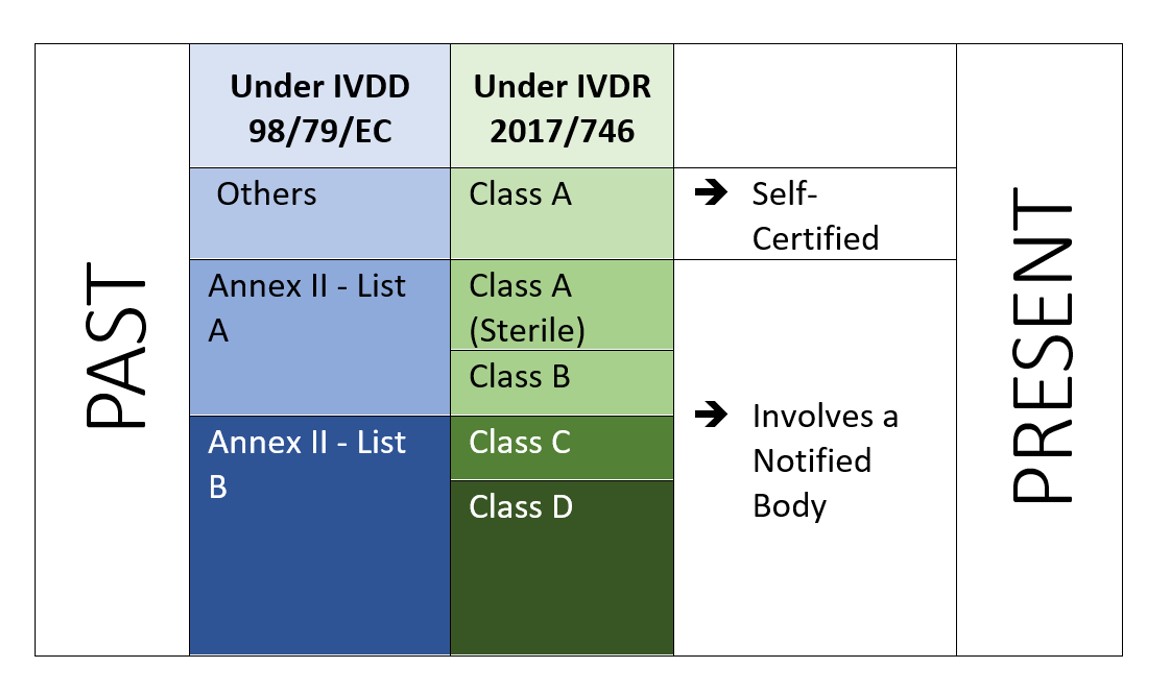
Figura 2: Classificazione basata sul rischio secondo IVDR 2107/746
3. Coinvolgimento dell'organismo notificato
Il ruolo di un organismo notificato (NB) sarebbe uno degli elementi centrali e quindi un numero maggiore di produttori avrebbe bisogno di essere controllato e certificato da un organismo notificato rispetto al metodo tradizionale di "autocertificazione".Gli operatori economici devono decidere con attenzione il percorso di valutazione della conformità (allegato IX, X, XI dell'EU IVDR[3]).
L'IVDR non solo richiede investimenti aggiuntivi, ma deve assicurare che la loro documentazione tecnica e il loro sistema di gestione della qualità soddisfino i nuovi requisiti dell'IVDR. Sotto la IVDD, la maggior parte degli IVD sono autocertificati (92%), e non richiedono il coinvolgimento di un organismo notificato (tranne 8% del totale degli IVD sul mercato[4]). Mentre con la nuova IVDR, lo scenario non è lo stesso.
Secondo uno studio "L'impatto delle nuove regole europee di classificazione degli IVD sul coinvolgimento dell'organismo notificato" a cura dell'Istituto Nazionale per la Salute pubblica and the Environment, Bilthoven (Netherlands) RIVM Letter report 2018-0082, A. van Drongelen et al., quasi 85% di tutti gli IVD richiederanno il coinvolgimento di un organismo notificato, lasciando solo 15% di IVD idonei all'autocertificazione[5]..
Questo significa anche che i produttori di diagnostica in vitro (IVD) dovranno affrontare un grande cambiamento per conformarsi al nuovo processo di classificazione e certificazione. Inoltre, a seconda dell'uso previsto dei dispositivi e della classe di rischio, il produttore deve identificare un NB designato che potrebbe essere in grado di controllarli e ottenere la certificazione dei loro prodotti. Il rischio più elevato IVD (Classe-D) richiederebbe un laboratorio di riferimento UE o gruppi di esperti per verificare le prestazioni dichiarate, oltre al coinvolgimento di un organismo notificato (NB) o di un'autorità competente (CA). Attualmente ci sono solo sei organismi notificati designati sotto l'IVDR dell'UE. Non aspettate a iniziare il vostro processo di applicazione per evitare ritardi imprevisti dovuti all'indisponibilità di un Organismo Notificato.

Figura 3: Elenco degli organismi notificati designati ai sensi dell'IVDR[6]
4. Istituzione di un sistema di gestione della qualità (QMS)
I produttori di IVD sono tenuti a stabilire un sistema di gestione della qualità (QMS) robusto e affidabile all'interno della loro sede. È un obbligo generale di un produttore ai sensi dell'articolo 10 dell'IVDR. Il sistema di gestione della qualità è un requisito essenziale tra vari altri, senza il quale un produttore non potrà essere approvato.
QMS is to ensure that manufacturing, change control, customer complaints, resource management, supplier &sub-contractors’ controls and validation, performance evaluation, quality test, UDI Labelling, Post market surveillance etc. are according to approved QMS and Post Market Surveillance (PMS) plans.
Il PRRC deve garantire che il produttore abbia soddisfatto i requisiti dell'articolo 10 per "autocertificare" (rilascio della dichiarazione di conformità secondo l'allegato IV) la classe A IVD quando un organismo notificato (NB) non è richiesto nel processo.
5. Essere preparati alle perturbazioni della catena di approvvigionamento
Throughout the world, manufacturer depends largely on their supply chain and raw material to produce and deliver IVDs that are safe, accurate, and effective forthe intended use. Hence regulatory and quality concerns are also evolving to a higher level when it comes to the suppliers and sub-contractors’ controls. Manufacturer are therefore expected to proactively communicate the supply chain about their obligations and responsibilities of the suppliers and subcontractors. Legal manufacturer shall demonstrate adequate supplier control and monitoring, assure the supply chain is in compliance to the regulatory aspects of IVDR, reconsider the need for data integrity and quality of supplier data, implement robust supplier risk management and performance monitoring and periodically audit the supplier based on the associated risk to the finished products. I regolatori e gli organismi notificati stanno enfatizzando i produttori legali per documentare chiaramente il livello dei controlli del fornitore e dimostrare con prove che hanno il potenziale per mitigare il rischio del prodotto o del servizio fornito dal fornitore.
6. Garantire la preparazione all'audit e all'ispezione
Secondo l'articolo 88 dell'IVDR, Attività di sorveglianza del mercato, le autorità competenti effettuano sia ispezioni annunciate (senza preavviso) nei locali degli operatori economici che dei fornitori e/o subappaltatori e, quando necessario, nelle strutture degli utenti professionali. Mentre il produttore deve includere le informazioni sull'identificazione di tutti i siti, compresi i fornitori e i subappaltatori, dove vengono eseguite le attività di produzione nella Documentazione Tecnica di Informazioni sulla Progettazione e la Produzione. Gli organismi notificati (NB) che eseguono l'audit del SGQ devono identificare i collegamenti tra i vari siti di produzione e i loro fornitori e/o subappaltatori e la ripartizione delle responsabilità. Queste informazioni saranno prese in considerazione quando l'NB vuole specificamente sottoporre ad audit uno di questi fornitori o subappaltatori o entrambi. I locali dei fornitori del fabbricante, quando si ritiene che influenzino significativamente la conformità dei dispositivi finiti, devono essere essenzialmente controllati dall'NB (in particolare quando il fabbricante non può dimostrare un controllo sufficiente sui suoi fornitori).
7. Piano per gestire gli audit non annunciati
Nell'ambito del monitoraggio post-certificazione, l'NB procede a verifiche in loco senza preavviso dei fabbricanti e dei loro subappaltatori o fornitori che eseguono prove sui prodotti e al monitoraggio della conformità con qualsiasi condizione che vincola i fabbricanti e associata alle decisioni di certificazione, come gli aggiornamenti dei dati clinici a intervalli definiti.Inoltre, tL'organismo notificato effettua a caso, almeno una volta ogni cinque anni, controlli senza preavviso sul sito del fabbricante e, se del caso, sul sito dei fornitori e/o subappaltatori del fabbricante, che possono essere combinati con la valutazione periodica di sorveglianza.
8. Rafforzare le attività di sorveglianza post-commercializzazione
Si raccomanda vivamente ai fabbricanti di rafforzare i loro requisiti di sorveglianza post-commercializzazione e di sviluppare un meccanismo di coordinamento tra gli Stati membri dell'UE in materia di vigilanza e sorveglianza del mercato. Nell'ambito della valutazione della sorveglianza applicabile ai dispositivi di classe C e D (allegato IX), l'organismo notificato esegue periodicamente, almeno una volta ogni 12 mesi, audit e valutazioni appropriate. Ciò comprende verifiche nei locali del fabbricante e dei fornitori e/o subappaltatori, a seconda dei casi. Il fabbricante sviluppa essenzialmente una procedura per la registrazione e la notifica degli incidenti e delle azioni correttive di sicurezza sul campo (FSCA).
9. Identificatore unico del dispositivo (UDI) & EUDAMED
Il produttore deve stabilire un sistema per l'UDI per identificare e facilitare la tracciabilità dei dispositivi. L'"identificatore del dispositivo" e l'"identificatore di produzione" devono essere presenti sulle etichette per migliorare la tracciabilità nel mercato UE. Si può fare riferimento a una lista di entità emittenti accreditate (IE) come GS1, HIBCC, ICCBBA, IFA GmbH per operare un sistema per l'assegnazione di UDI. Attualmente gli IE menzionati sono validi dal 27il Giugno 2019, ma sarà saggio confermare la loro validità mentre si prende una decisione finale sulla loro attuazione.
European Database on Medical Devices (EUDAMED) fornirà una panoramica di tutti i dispositivi medici disponibili nell'Unione Europea. Consiste di sei moduli relativi a:
- Registrazione dell'attore,
- Identificazione unica del dispositivo (UDI) e registrazione del dispositivo,
- Organismi notificati e certificati,
- Indagini cliniche e studi di performance,
- Vigilanza e sorveglianza post mercato, e
- Sorveglianza del mercato.
Per garantire una maggiore trasparenza attraverso un EUDAMED completo, parti delle informazioni degli operatori economici saranno accessibili al pubblico. Mentre le informazioni riservate saranno accessibili solo all'Operatore Economico, agli Sponsor, alle Autorità Notificate e Competenti degli stati membri dell'UE.
10. Requisiti per i "dispositivi interni"
Health institution developing ‘in-house devices’ (or ‘laboratory-developed tests’) which are meant to be used by the same health institution shall not be marketed or sold to other legal entity. Such devices may be used for the diagnosis and treatment, especially for rare diseases. The institution is expected to comply with only the requirement of Annex I of IVDR (general safety and performance requirements), and exempted from rest of the regulation until 26 May 2024; provided the health institution meets a number of conditions set out in Article 5(5) of the Regulation and has an appropriate quality management system, which complies to the international standard setting out the quality and competence requirements for medical laboratories (EN ISO 15189) or other national provisions, and is able to justify that target patient group’s specific needs cannot appropriately be met by an equivalent device available on the market.
Riferimenti
[1] Comunicato stampa ufficiale della CE del 20il Dicembre 2021, introduzione progressiva del regolamento sui dispositivi medici diagnostici in vitro. Si può accedere a https://ec.europa.eu/commission/presscorner/detail/en/IP_21_6965 [2]REGOLAMENTO (UE) 2017/746 DEL PARLAMENTO EUROPEO E DEL CONSIGLIO del 5 aprile 2017 relativo ai dispositivi medico-diagnostici in vitro e che abroga la direttiva 98/79/CE e la decisione 2010/227/UE della Commissione; ALLEGATO VIII REGOLE DI CLASSIFICAZIONE, 1. REGOLE DI ATTUAZIONE Punto 1.4 Pagina 304 [3]Regolamento (UE) 2017/746 del Parlamento europeo e del Consiglio del 5 aprile 2017 sui dispositivi medico-diagnostici in vitro e che abroga la direttiva 98/79/CE e la decisione 2010/227/UE della CommissioneALLEGATO IX Valutazione della conformità basata su un sistema di gestione della qualità e sulla valutazione della documentazione tecnica, pagina 306, ALLEGATO X Valutazione della conformità basata sull'esame del tipo, pagina 314, ALLEGATO XI Valutazione della conformità basata sulla garanzia della qualità della produzione, pagina 317
[4] Comunicato stampa dtd. 14 ottobre 2021, Bruxelles; Salute pubblica: La Commissione propone un'introduzione progressiva del nuovo regolamento sui dispositivi medici diagnostici in vitro [5]L'impatto delle nuove regole europee di classificazione IVD sul coinvolgimento dell'organismo notificato: uno studio sugli IVD registrati nei Paesi Bassi; van Drongelen A, de Bruijn A, Pennings J, van der Maaden T 32 p in inglese 2018, rapporto lettera RIVM 2018-0082 [6] L'elenco di cui sopra si basa su dati accessibili dtd. 5 marzo 2021, per gli ultimi aggiornamenti sulla lista, è possibile accedere al sito ufficiale della CE all'indirizzo https://ec.europa.eu/growth/tools-databases/nando/index.cfm?fuseaction=directive.notifiedbody&dir_id=35Kolabtree helps businesses worldwide hire freelance scientists and industry experts on demand. Our freelancers have helped companies publish research papers, develop products, analyze data, and more. It only takes a minute to tell us what you need done and get quotes from experts for free.

Unlock Corporate Benefits
• Secure Payment Assistance
• Onboarding Support
• Dedicated Account Manager
Sign up with your professional email to avail special advances offered against purchase orders, seamless multi-channel payments, and extended support for agreements.


