



Presentazioni FDA consulente e scrittore di normative Samradni Patil fornisce un Lista di controllo delle presentazioni 510k to help dispositivo medico aziende con un'autorizzazione FDA facile e veloce.
Il processo di presentazione 510(k) è usato tipicamente per i dispositivi medici di Classe II per ottenere l'autorizzazione dalla US Food and Drug Administration (FDA). Il processo Premarket Approval (PMA) è usato di solito per i dispositivi medici di Classe III.
Il processo di revisione 510(k) determina l'equivalenza sostanziale (SE) con un dispositivo simile legalmente commercializzato, chiamato anche dispositivo predicato. Il dispositivo deve essere sicuro ed efficace almeno quanto il dispositivo legalmente commercializzato per affermare che è sostanzialmente equivalente ad esso. Il dispositivo sotto esame 510(k) deve mostrare quanto segue per rivendicare la SE con il dispositivo predicato:
- Stessa destinazione d'uso del dispositivo legalmente commercializzato (dispositivo predicato) predicato
- Le stesse caratteristiche tecnologiche del dispositivo predicato o
- Diverse caratteristiche tecnologiche e informazioni/test che suggeriscono che il dispositivo è sicuro ed efficace come il dispositivo predicato e diverse questioni di sicurezza ed efficacia rispetto al dispositivo predicato non vengono sollevate.
Il mancato rispetto dei criteri di cui sopra porta alla determinazione di Non-Substantial Equivalence (NSE).
Lista di controllo delle presentazioni 510k
Il processo di revisione FDA 510(k) può essere ampiamente diviso in 2 fasi.
- Revisione dell'accettazione
- Revisione sostanziale
Revisione delle scadenze
| Tipo di recensione | Tempistica (giorni di calendario) | Risultato del processo |
| Revisione dell'accettazione | Entro il 15° giorno | La FDA informa il richiedente se la domanda è accettata per Revisione sostanziale o Messo in attesa RTA |
| Revisione sostanziale | Entro il giorno 60 | Recensione interattiva o Richiesta di informazioni aggiuntive |
Nota: il giorno 1 è il giorno in cui la FDA riceve la domanda 510(k)
Discutiamo prima quali tipi di problemi vengono affrontati dalle aziende di dispositivi medici durante questi processi di revisione.
Revisione dell'accettazione
Se la 510(k) non viene accettata in questa fase, viene messa su Rifiutare di accettare (RTA) Hold. Come per Dati FDANel 2018 circa 30% 510(k) sono stati messi in attesa di RTA.
Revisione sostanziale
Il grafico sottostante mostra la percentuale di richieste di informazioni aggiuntive emesse dalla FDA durante la fase di revisione sostanziale.
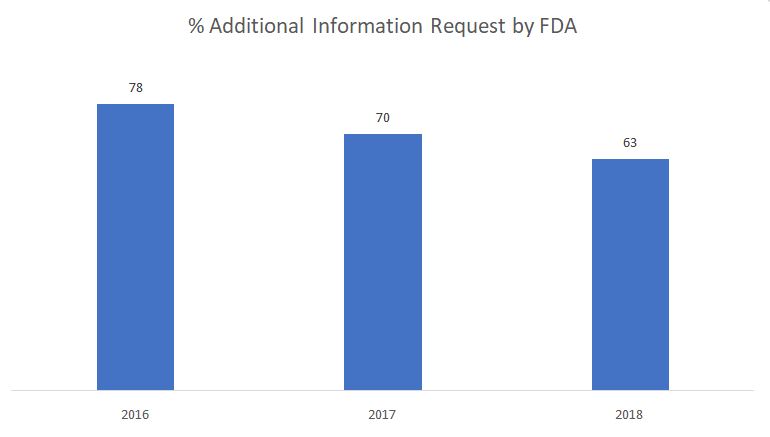
Fonte: FDA
La percentuale di 510ks determinata come Non sostanzialmente equivalente (NSE) è mostrato qui sotto.
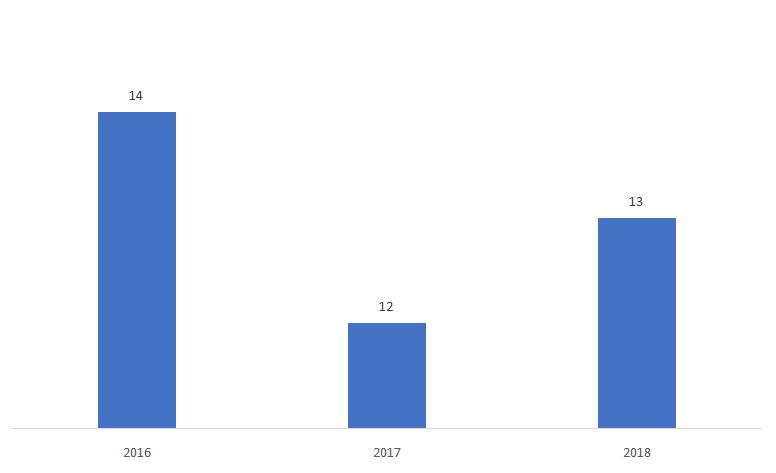
Fonte: FDA
Richieste di informazioni aggiuntive comuni della lista di controllo della presentazione 510k
Ora che abbiamo capito le questioni comuni sollevate come parte del processo di revisione 510k, concentriamoci su quali tipi di richieste di informazioni aggiuntive sono comuni.
Il Dati FDA mostra i seguenti tipi di richieste di informazioni aggiuntive:
- Descrizione inadeguata del dispositivo
- Discrepanze nella presentazione - Le discrepanze in questa categoria sono più spesso legate alla descrizione del dispositivo o alle indicazioni d'uso
- Problemi con le indicazioni d'uso
- Non seguire o affrontare in altro modo gli attuali documenti guida o gli standard riconosciuti
- Manca completamente il test delle prestazioni richiesto per alcuni tipi di dispositivi (cioè, non vengono forniti dati sulle prestazioni)
- I dati clinici richiesti per alcuni tipi di dispositivi sono completamente mancanti (cioè, nessun dato clinico fornito)
Ora, discutiamo le migliori pratiche da seguire durante la preparazione e la presentazione di una domanda 510k.
Lista di controllo per la presentazione 510k:
1. Lettera di rifiuto dell'accettazione (RTA)
Lo scopo della revisione dell'accettazione nella fase iniziale è quello di controllare se la domanda 510k è amministrativamente completa. Si raccomanda vivamente di rivedere la lista di controllo di accettazione fornita nel documento guida "Rifiuto di accettare la politica per 510k".
Per garantire il successo della revisione di accettazione, si suggerisce ad ogni azienda di seguire gli elementi della seguente tabella dal documento di orientamento.
- Tabella delle domande preliminari: Anche se questa lista di controllo è destinata al revisore principale per fare la determinazione iniziale, si raccomanda vivamente di rispondere a queste domande in modo informale prima di presentare la domanda alla FDA.
- Tabella degli elementi organizzativi: Questi elementi aiutano a organizzare 510(k) in un modo che permette una facile identificazione delle informazioni nell'applicazione 510(k).
- Elementi di una presentazione completa (articoli RTA) Tabella: Le aziende dovrebbero prestare maggiore attenzione agli elementi elencati in questa tabella poiché questi elementi sono critici per non ottenere una lettera di RTA.
2. Descrizione inadeguata del dispositivo
La descrizione del dispositivo è obbligatoria nella domanda 510(k). Si suggerisce di aggiungere una breve descrizione e le specifiche tecniche in questa sezione. Tutti i modelli di dispositivi medici e gli accessori dovrebbero essere inclusi. Immagini, diagrammi, dimensioni, disegni e tolleranze per ogni componente dovrebbero essere inclusi. La mancanza di un modello o di un accessorio importante può portare a confusione e a ulteriori domande. Specifiche tecniche inadeguate possono portare a un malinteso e alla richiesta di ulteriori test.
3. Informazioni incoerenti in tutta la presentazione
- Incoerenza nella descrizione del dispositivo: Se un'azienda decide di fare la presentazione 510(k) per aggiungere un modello aggiuntivo, è importante che le sezioni applicabili come la lettera di accompagnamento, la descrizione del dispositivo, l'etichettatura, la discussione sull'equivalenza sostanziale, le sezioni relative alle prestazioni siano allineate al cambiamento effettivo. L'incoerenza può portare a ritardi amministrativi e nel peggiore dei casi alla richiesta di test aggiuntivi.
- Incoerenza nelle indicazioni d'uso: Simile alla descrizione del dispositivo, l'incoerenza nell'indicazione d'uso in varie sezioni del 510(k) può causare problemi. La dichiarazione di indicazione d'uso è molto importante per fare la determinazione SE.
Le incongruenze in varie sezioni potrebbero essere facilmente evitate con un'attenta revisione della presentazione prima che sia presentata alla FDA. È sempre una buona idea avere un paio di occhi in più per dare un'occhiata alle varie sezioni per evitare tali errori.
4. Diversa destinazione d'uso rispetto al dispositivo predicato
Per ottenere la determinazione SE dalla FDA, il dispositivo deve avere il stessa destinazione d'uso del dispositivo predicato. Questo è importante perché la diversa destinazione d'uso rispetto al dispositivo predicato può portare a diversi problemi di sicurezza ed efficacia. In tal caso, 510(k) potrebbe non essere un percorso appropriato per ottenere l'autorizzazione del prodotto. Va notato che le differenze nell'indicazione d'uso tra il dispositivo predicato e il dispositivo sottoposto a revisione 510(k) possono non risultare necessariamente in una diversa destinazione d'uso. Le aziende dovrebbero fare ulteriori sforzi per dimostrare chiaramente che le differenze nell'indicazione d'uso non hanno portato a una diversa destinazione d'uso.
La guida della FDA "Il programma 510(k): Valutare l'equivalenza sostanziale nelle notifiche pre-commercializzazione [510(k)]" dovrebbe essere riferito per ottenere chiarezza sulle domande relative all'uso previsto.
5. Informazioni inadeguate/mancanti sui test
- Informazioni inadeguate sui test:
È importante capire i test applicabili per ogni particolare dispositivo. In base al tipo di dispositivo, possono essere richiesti test di sicurezza elettrica, test di compatibilità elettromagnetica (EMC), test di biocompatibilità, test di validazione del software, test di sterilizzazione, test di usabilità per rivendicare la sicurezza e l'efficacia di un dispositivo.
Spesso, le aziende sottovalutano la quantità di test richiesti o cercano di fornire motivazioni per non condurre alcun test particolare
Esempio: Le aziende possono basarsi sui dati di biocompatibilità di un dispositivo simile per dichiarare la biocompatibilità del loro prodotto. Questo approccio può essere accettabile in alcuni casi. Tuttavia, molte volte il processo di produzione tra questi dispositivi può giustificare test di biocompatibilità separati sul dispositivo sotto la revisione 510(k).
Questi test aggiuntivi possono richiedere diverse settimane. Se la FDA richiede di condurre questi test aggiuntivi durante la revisione 510(k), si aggiunge molto tempo nella finale 510(k) di autorizzazione.
Appropriate teams should give thorough consideration to current FDA guidance, product design, risk management process to make determination about amount of testing required.
- Informazioni mancanti sui test:
È importante capire la differenza tra la 510(k) tradizionale e la 510(k) speciale. Tutti i dati dei test devono essere inclusi nella presentazione tradizionale 510(k).
6. Omissione di seguire o altrimenti affrontare Corrente Documento/i di guida o standard riconosciuti
Come discusso in precedenza, assicurarsi che i test siano condotti per dimostrare la conformità agli ultimi standard riconosciuti. Ci possono essere cambiamenti significativi nell'ultima versione dello standard rispetto alla versione precedente. Questo può comportare ulteriori domande sulla sicurezza e l'efficacia se il dispositivo non è testato con l'ultima versione dello standard.
Fare riferimento agli ultimi documenti guida aiuta a capire le aspettative o le raccomandazioni della FDA. Questo rende anche il processo di revisione della FDA facile perché la presentazione è scritta in un formato facilmente comprensibile per il revisore.
7. Determinazione NSE
L'obiettivo finale del processo 510(k) è quello di determinare l'equivalenza sostanziale (SE) con un dispositivo predicato. Quando la FDA richiede ulteriori informazioni durante la fase di revisione sostanziale, le aziende devono esaminare attentamente ogni richiesta e fornire una risposta scientificamente valida. La mancata fornitura dei dati/risposta richiesti può portare alla determinazione di NSE. Molte determinazioni dell'NSE sono dovute alla mancanza di fornire dati sulle prestazioni.
Si consiglia vivamente di consultare e collaborare con la FDA per capire le aspettative in questa fase. Ecco alcuni altri errori comuni che ho notato nella mia esperienza.
Aspetti amministrativi
8. Presentazione della domanda all'indirizzo corretto
Questo è un errore umano facilmente evitabile. Fate sempre riferimento al sito web della FDA per il indirizzo corretto per presentare la sua domanda.
9. Compresa la copia cartacea e la copia elettronica come da ultima raccomandazione della FDA
La FDA ha requisiti specifici per il numero di copie cartacee ed elettroniche che devono essere presentate per l'applicazione 510(k). Attualmente, è richiesta 1 copia cartacea e 1 copia elettronica. Non fare supposizioni. Fate riferimento al sito web della FDA prima di saltare a qualsiasi conclusione.
10. problemi relativi a eCopy
La presentazione deve soddisfare gli standard tecnici di eCopy. Fare riferimento al Guida eCopy per preparare la copia elettronica della domanda.
La convenzione di denominazione dei PDF e la raccomandazione sulla dimensione del file devono essere seguite per evitare la lettera di sospensione di eCopy. Anche se l'uso del Strumento eSubmitter-eCopie è volontario, questo strumento aiuta a convalidare l'eCopy secondo le raccomandazioni della FDA.
11. Presentazione delle informazioni al revisore
Dopo aver ricevuto la lettera iniziale di sospensione da parte dell'FDA (RTA hold, eCopy hold), le aziende spesso commettono un errore nel presentare le informazioni al revisore. Controllare l'e-mail ricevuta dalla FDA o la guida appropriata della FDA su dove inviare la risposta alla lettera di sospensione.
Aspetti tecnici
12. 510(k) tradizionale vs 510(k) speciale
Le aziende possono commettere un errore nel classificare l'applicazione come 510(k) tradizionale o 510(k) speciale. La differenza principale tra la 510(k) tradizionale e la 510(k) speciale è il tempo richiesto per la revisione della domanda da parte della FDA. La 510(k) speciale richiede 30 giorni di calendario mentre la 510(k) tradizionale richiede 90 giorni di calendario. Ho visto diverse volte la FDA richiedere alle aziende di convertire la Special 510(k) in Traditional 510(k). In questo caso, le aziende finiscono per perdere molto tempo nel processo di conversione.
Secondo la FDA, una 510(k) speciale può essere appropriata quando:
- Il cambiamento proposto è presentato dal fabbricante legalmente autorizzato a commercializzare il dispositivo esistente;
- I dati di performance non sono necessari, o se i dati di performance sono necessari, sono disponibili metodi ben stabiliti per valutare il cambiamento; e
- Tutti i dati sulle prestazioni necessari a sostenere l'equivalenza sostanziale possono essere esaminati in un formato di sintesi o di analisi del rischio.
To avoid such mistake, do thorough ricerca on the FDA database to identify if similar change was submitted as Special 510(k) or Traditional 510(k). Refer to the documenti di orientamento dalla FDA. Se siete ancora in dubbio, fatevi aiutare da consulenti normativi. Usare un approccio basato sul rischio. Se avete ancora dei dubbi, si raccomanda di adottare un approccio conservativo e di presentare una 510(k) tradizionale.
Sfide uniche
13. Natura del dispositivo
Alcuni dispositivi possono sollevare questioni uniche a causa della natura unica del dispositivo. Alcune aree tecnologiche come Intelligenza artificiale (AI) e Cybersecurity sono relativamente nuovi. La FDA ha collaborato con l'industria per sviluppare documenti guida in queste aree.
In questi casi, è altamente raccomandato un incontro preliminare con la FDA prima della presentazione della 510(k). Fare riferimento al Documento guida della FDA se le aziende hanno bisogno di un feedback e di un incontro con la FDA prima della presentazione della 510(k).
Conclusione
Questi erano i punti importanti in una lista di controllo della presentazione 510(k). Le lettere di Refuse to Accept (RTA) da parte della FDA potrebbero essere facilmente evitate con un'attenta revisione della presentazione e seguendo i documenti guida della FDA.
La richiesta di informazioni aggiuntive come parte del processo di revisione sostanziale potrebbe essere ridotta scrivendo 510(k) chiari e concisi. Questo è spesso una combinazione di scienza, arte ed esperienza. Si raccomanda vivamente di prendere l'aiuto di esperti Consulenti in materia di regolamentazione nella scrittura di 510(k) per evitare errori costosi, seguite questa lista di controllo per la presentazione 510(k). Rimanete aggiornati in termini di regolamenti, standard e documenti guida applicabili per aumentare le possibilità di una presentazione di successo.
Necessità di consultare un Esperto in presentazioni FDA? Lavorare con esperti redattori di normative, esperti dell'industria dei dispositivi medici e Consulenti 510k che hanno aiutato le aziende di dispositivi medici a preparare i documenti normativi per ottenere l'autorizzazione della FDA.
Kolabtree helps businesses worldwide hire freelance scientists and industry experts on demand. Our freelancers have helped companies publish research papers, develop products, analyze data, and more. It only takes a minute to tell us what you need done and get quotes from experts for free.

Unlock Corporate Benefits
• Secure Payment Assistance
• Onboarding Support
• Dedicated Account Manager
Sign up with your professional email to avail special advances offered against purchase orders, seamless multi-channel payments, and extended support for agreements.


