



Shrinidh Joshi, expert en dispositifs médicaux on Kolabtree, provides a comprehensive guide to dispositif médical design, design controls, validation & verification, regulatory requirements and risk management.
Dans le article précédent, we took a look at the overview of the dispositif médical development process from the ideation to the discovery phase. In this article, we will focus more on medical device design, design controls, and compliance.
Conception de dispositifs médicaux : Réglementations et conformité IEC & ISO
Vous savez maintenant que pour être mis sur le marché, votre dispositif médical doit répondre à certaines exigences et normes réglementaires. Les normes relatives aux dispositifs médicaux, telles que celles de la Commission électrotechnique internationale (CEI) ou de l'Organisation internationale de normalisation (ISO), permettent aux fabricants de dispositifs médicaux, aux concepteurs, aux laboratoires et à tous les autres prestataires de services de développement de dispositifs médicaux, tels que les CDMO, d'inspecter, d'évaluer et de maintenir leurs dispositifs et équipements selon certaines normes de qualité et d'utilisation.
La CEI a publié la première norme de ce type sur les dispositifs médicaux en 1970, IEC 60601-1. CEI 60601-1, Appareils électromédicaux - Partie 1 : C'est la norme internationalement reconnue qui traite des exigences générales pour les appareils électromédicaux et les dispositifs couvrant les normes de sécurité de base et les performances essentielles. [4].
Le document CEI 60601-1 a été révisé périodiquement pour rester en phase avec les derniers développements médicaux et les avancées technologiques dans le domaine des dispositifs médicaux. Le changement le plus récent est intervenu en 2012 (Amendement 1 à la CEI 60601-1). Cette norme révisée contient les exigences relatives à la prise en compte du facteur humain, à l'évaluation des performances essentielles des dispositifs médicaux, à l'utilisabilité et aux commandes. Elle inclut également les logiciels en tant que dispositifs médicaux et spécifie leur adoption d'un cycle de vie de développement formel. La norme CEI 60601-1 révisée comprend également des spécifications techniques nouvelles et révisées concernant les risques (électriques et mécaniques), les exigences en matière d'étiquetage des dispositifs médicaux (y compris les nouvelles normes d'étiquetage) et la documentation.
Conception de dispositifs médicaux : Normes ISO
L'Organisation internationale de normalisation dispose également de spécifications pour les normes relatives aux dispositifs médicaux. ISO 13485 et ISO 14971 sont des normes largement utilisées dans le monde entier pour la gestion de la qualité des dispositifs médicaux. Outre ces normes internationales, certaines normes sont spécifiques à une région et toutes sont adoptées à partir des normes internationales avec quelques modifications et limitations.
Si une entreprise de dispositifs médicaux fabrique ou vend des dispositifs médicaux aux États-Unis, ceux-ci seront réglementés par la FDA. L'American National Standards Institute (ANSI) est le représentant des normes ISO aux États-Unis.
Il existe deux autres organisations similaires : l'Association for the Advancement of Medical Instrumentation (AAMI) et l'American Society for Quality (ASQ) qui définit les normes pour les États-Unis.
Si une entreprise de dispositifs médicaux a conçu un dispositif en tenant compte des normes ISO, il est également possible que la FDA n'approuve pas le dispositif. En effet, la FDA dispose de son propre ensemble de procédures de gestion des risques dérivées des normes internationales et régionales, qui comprennent :
(norme internationale.)
- ANSI/AAMI/ISO 14971:2007 (R2010), Dispositifs médicaux - Application de la gestion des risques aux dispositifs médicaux. (Une norme régionale avec des ajouts et des modifications par rapport à la norme internationale visée). [5].
Dans le cas de la norme de gestion de la qualité, elle ne suit pas la version internationale ou régionale de la norme ISO 13485. Cela s'explique par le fait que la FDA a des directives différentes en matière de gestion de la qualité des dispositifs médicaux pour le marché américain.
En revanche, si l'entreprise de dispositifs médicaux envisage l'Union européenne, le Comité européen de normalisation (CEN) est la normalisation adoptée par l'ISO et le Comité européen de normalisation électrotechnique (CENELEC) est la norme régionale inspirée par la CEI.
Le CEN est un peu modifié selon les exigences de l'ISO et s'écrit avec le préfixe "EN". Par exemple :
- EN ISO 13485:2012, Dispositifs médicaux - Systèmes de management de la qualité - Exigences à des fins réglementaires.
- EN ISO 14971:2012, Dispositifs médicaux - Application de la gestion des risques aux dispositifs médicaux.
Les membres nationaux adoptent ces normes de l'UE en ajoutant leur préfixe. Pour la Suisse, Swiss Standards publie des normes avec le préfixe "SN", comme SN EN ISO 13485:2012 et SN EN ISO 14971:2012.
Dans le cas du Canada, l'Autorité canadienne de normalisation (CSA) est l'organisme qui représente l'ISO.
Réglementation des dispositifs médicaux et contrôle de la conception
Les fabricants de dispositifs médicaux doivent suivre les directives de contrôle de la conception car les organismes de réglementation comme la FDA, la Commission européenne, Santé Canada et d'autres veulent s'assurer que les dispositifs médicaux sont sûrs pour les utilisateurs potentiels avant que les fabricants ne commencent à les commercialiser. Comme je l'ai mentionné dans la section ci-dessus, la FDA ne suit pas la norme ISO 13485 car elle a des exigences différentes en matière de gestion de la qualité. Les contrôles de conception sont définis sous FDA 21 CFR 820.30 qui a une intention similaire à la section 7.3 Conception et développement décrite dans les lignes directrices de la norme ISO 13485. En outre, la FDA intègre les exigences des bonnes pratiques de fabrication actuelles (cGMP) dans le règlement sur les systèmes de qualité afin de suivre les bonnes pratiques de qualité pour la conception des dispositifs médicaux. [6].
Le règlement fournit un cadre permettant de mettre en œuvre le contrôle de la conception pour une grande variété de dispositifs. Ce cadre offre une certaine souplesse, tant pour la mise en conformité avec la réglementation que pour le processus interne de conception et de développement.
Pour réussir à mettre en œuvre le contrôle de la conception des dispositifs médicaux, il faut des professionnels ayant une formation à la fois technique et non technique, comme l'administration des affaires, les sciences de la vie, l'ingénierie, l'informatique et les arts.
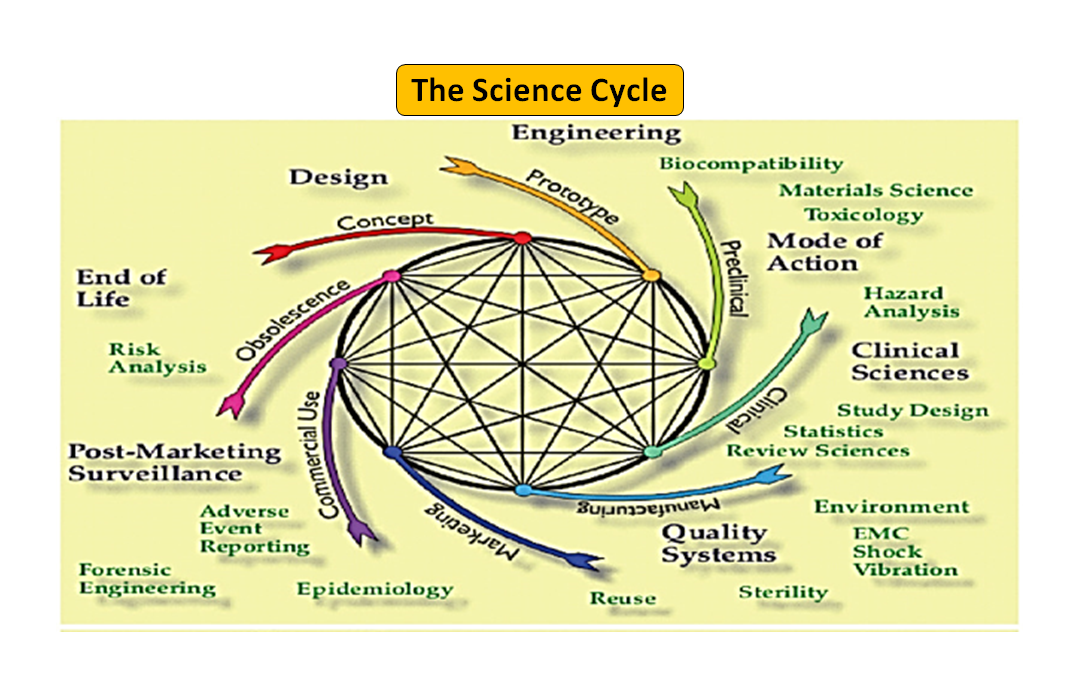
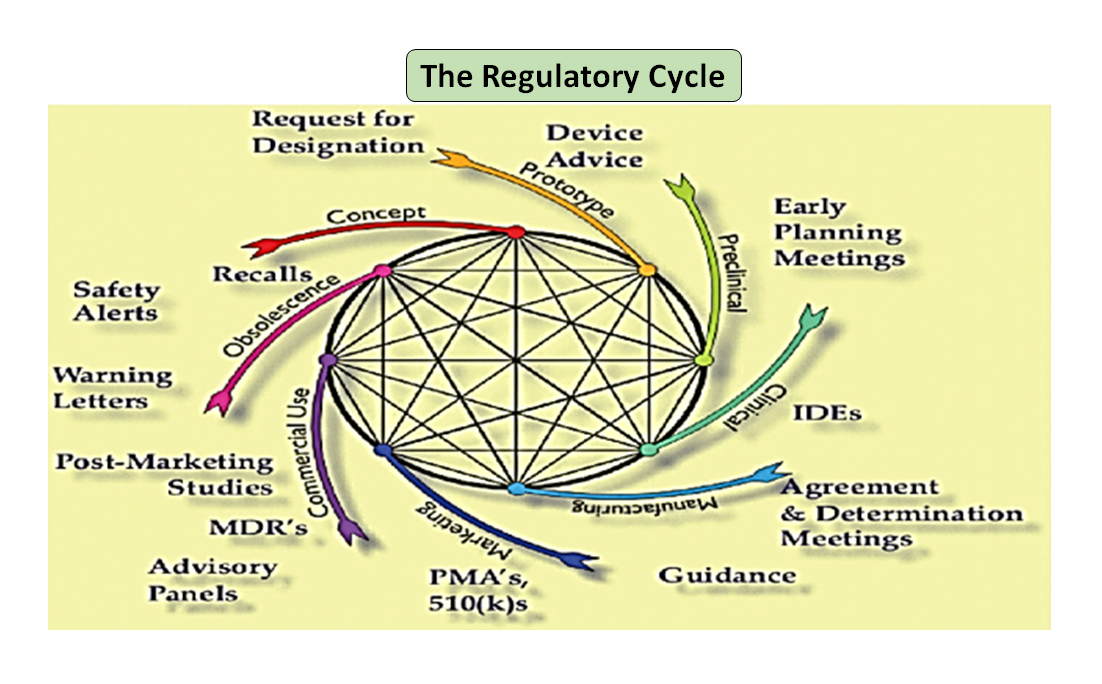
Figure 1 : Le cycle de vie total du produit. Le cycle scientifique et le cycle réglementaire (Adapté de [7]).
Il convient de noter que le cycle de vie d'un dispositif médical, de l'innovation à l'approbation réglementaire et à la commercialisation, est une série d'étapes interconnectées qui déterminent le développement du dispositif (voir la figure 1 : cycle total du produit). Au début, les prototypes conçus par vos ingénieurs sont testés sur banc d'essai afin d'optimiser la conception, de vérifier la biocompatibilité, les matières extractibles, les matières lixiviables, la flexibilité ou la résistance globale de votre dispositif. Le rôle du conseiller réglementaire de votre entreprise est de parcourir la base de données réglementaire pour vous suggérer un document d'orientation qui peut vous aider à déterminer si votre produit sera réglementé en tant que dispositif médical ou non. L'utilisation prévue de votre dispositif médical et son mode de fonctionnement ou d'action vous guideront dans la conception du dispositif et décideront également de la voie réglementaire à suivre, qu'il s'agisse de 510(k), PMA, De Novo, Pre-sub, IDE, HDE, fichiers maîtres, etc.
Comme l'illustre la figure 1, les processus scientifiques et réglementaires sont imbriqués tout au long du cycle de vie du produit. Tout comme les différentes parties du cycle de vie scientifique sont interconnectées, la science et les exigences réglementaires sont entrelacées, chacune informant et déterminant l'autre. Il est possible de créer des liens, tant à la FDA que chez les fabricants, afin que certaines parties du cycle de vie ne risquent pas d'être considérées de manière isolée. Par exemple, il n'est pas rare qu'une demande de précommercialisation soit examinée sans tenir compte de l'expérience post-commercialisation de produits similaires.
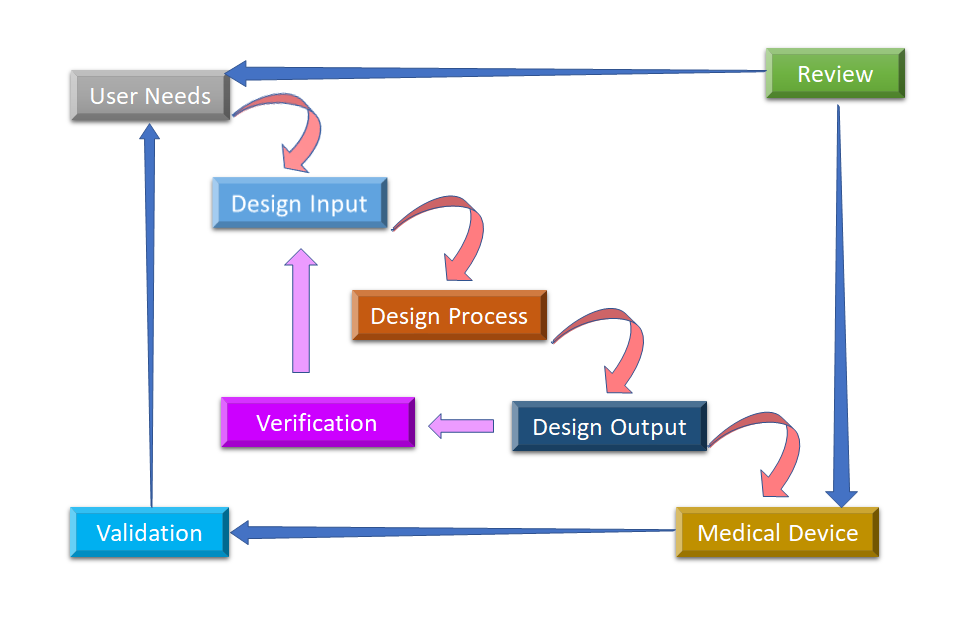
Figure 2 : Processus de conception du flux d'eau pour le contrôle de la conception des dispositifs médicaux (Adapté de [8]).
La phase initiale à partir de laquelle commence le contrôle de la conception est le développement et l'approbation de l'entrée de conception, qui consiste en la conception du dispositif et les processus de fabrication à réaliser dans la phase de production. Le contrôle de la conception est une approche holistique qui ne se limite pas au transfert de la conception vers la phase de production, une fois la conception finalisée. Il influe également sur les processus de fabrication en fonction des modifications apportées en phase de conception ou même du retour d'information post-production. Le développement d'un produit utilisable par l'utilisateur est un processus continu. Ainsi, pour le produit amélioré, il prend en compte les changements révolutionnaires issus des modèles d'utilisation ainsi que l'analyse des produits ayant échoué. Vous pouvez voir à la figure 2 comment le contrôle de la conception peut être effectué dans le processus de conception en cascade.
- Besoins des utilisateurs:- Les exigences sont définies en tenant compte du besoin du marché et le dispositif est conçu pour répondre à ce besoin. Après une série d'évolutions, la conception du dispositif médical est finalisée et transférée à la production pour la fabrication. Un retour d'information est nécessaire à chaque étape de ce processus.
- Design Input : Il s'agit d'un processus itératif. Lorsqu'une organisation décide de répondre à un besoin particulier, elle examine et teste l'acceptabilité des données de conception dérivées de ce besoin. C'est alors que commence le processus itératif de conversion des exigences en conception de dispositifs.
- Processus de conception : Ces entrées de conception sont converties en sorties de conception en convertissant ces exigences en spécifications de haut niveau (qui sont des sorties de conception).
- Design Output : Le processus de vérification confirme si les spécifications satisfont ou non les exigences. La sortie devient l'entrée pour réviser les exigences et ce processus se poursuit jusqu'à ce que la sortie de conception soit alignée avec l'entrée de conception.
- Dispositif médical : Once the final design is ready, it is transmitted to the production facility for mass manufacturing. Design control regulation mandates Design History File (DHF), which illustrates the linkages and relationships between all the Design Controls and help to trace all changes throughout the entire développement de produits processus.
Les entreprises de dispositifs médicaux peuvent adopter une approche papier ou logicielle, spécialement développée pour le contrôle de la conception ; les fichiers de l'historique de la conception doivent être traçables et accessibles à tous les membres de l'équipe.
L'organigramme ci-dessous présente une étude de cas pour le contrôle de la conception d'un dispositif médical.

Conception de dispositifs médicaux : Pourquoi la traçabilité est importante
Currently in the realm of the medical devices industry, it is an ideal practice to develop a traceability matrix that can illustrate the links and relations between user needs, design inputs and outputs, design verification and validation. When you are in the early phase for your device development you can maintain the device traceability using a spreadsheet or document version but as you move forward, its good idea to use cloud-based project management and document sharing platforms such as Microsoft Teams, Asana, Trello or whichever platform is suitable for your organization. The goal is as your project progresses you need to find an option which can save time because the old-school method of maintaining a traceability matrix might consume a lot of your time which you should rather be focusing on design verification and validation.
Une matrice de traçabilité des contrôles de conception est essentielle pour les équipes de développement de produits, et en particulier pour les chefs de projet, car la traçabilité montre la relation et les liens entre tous les contrôles de conception. Comment les besoins de l'utilisateur sont-ils liés aux intrants de la conception ? Comment les résultats de la conception sont-ils liés aux données d'entrée de la conception ? Comment les vérifications de la conception sont-elles liées aux intrants et aux extrants de la conception ? Comment les validations de conception sont-elles liées aux besoins de l'utilisateur ? Une matrice de traçabilité est un outil précieux pour montrer une vue de haut niveau et le flux du développement d'un produit de dispositif médical du début à la fin.
Les développeurs de produits aux pratiques exemplaires s'appuient sur la traçabilité des contrôles de conception depuis de très nombreuses années. Et maintenant ISO 13485:2016 fait également de la traçabilité une exigence. Comme cité dans la norme ISO 13485:2016, 7.1 Planification de la réalisation du produit, 1. c) les activités requises de vérification, de validation, de surveillance, de mesure, d'inspection et d'essai, de manipulation, de stockage, de distribution et de traçabilité spécifiques au produit, ainsi que les critères d'acceptation du produit ; et 7.3.2 Planification de la conception et du développement, 1. e) les méthodes permettant d'assurer la traçabilité des sorties de conception et de développement par rapport aux entrées de conception et de développement. [9].
Conception de dispositifs médicaux : Vérification et validation
Chaque dispositif médical doit répondre aux objectifs de fonctionnalité, de convivialité et de fiabilité pour s'imposer sur le marché.
En outre, vos parties prenantes (patients, prescripteurs, régulateurs ou utilisateurs finaux) seront également attentives à la sécurité et à l'efficacité de votre dispositif. Il est très probable que votre dispositif soit conçu pour répondre à un besoin non satisfait qui peut être essentiel à la vie, par exemple un ventilateur ou un dispositif de diagnostic qui peut détecter une maladie cardiaque. Par conséquent, les tests itératifs de votre dispositif, avec vérification et validation, sont cruciaux. Ces deux étapes du processus de conception visent à confirmer que votre dispositif médical est conforme aux exigences des utilisateurs et qu'il fonctionne comme prévu. En termes simples, la vérification et la validation de la conception peuvent garantir que votre dispositif fait réellement ce qu'il est censé faire. La vérification et la validation de la conception permettent également de garantir les exigences réglementaires, les normes, la qualité du produit et le processus de fabrication de votre dispositif médical. La vérification de la conception permet d'évaluer si le résultat de votre conception est conforme aux exigences spécifiées, aux spécifications ou aux exigences réglementaires qui sont spécifiées dans l'entrée de la conception. D'autre part, la validation de la conception est destinée à évaluer si votre dispositif médical apporte des avantages en fonction des besoins des utilisateurs finaux.
Design vérification demande : "Avons-nous bien conçu l'appareil ?"
Design validation demande : "Avons-nous conçu le bon appareil ?"
Les dispositifs médicaux peuvent être constitués de différentes formes technologiques, de différentes tailles et de différents niveaux de complexité. L'activité de vérification et de validation (V&V) est déterminée par l'environnement réglementaire et doit suivre les normes internationales. Les activités normalisées de V&V peuvent rationaliser le processus de fabrication et améliorer le processus d'approbation. En outre, les tests automatisés, les techniques de diagnostic et les outils de collecte de données peuvent améliorer le processus de V&V. [10].
- Validation du produit vs. validation du processus
- Conception du dispositif médical/validation du produit : - Conformité aux besoins de l'utilisateur et du patient, c'est-à-dire : le dispositif fonctionne-t-il correctement ?
- Validation du processus : le processus de fabrication répond à des spécifications prédéterminées.
Il convient de rappeler que la validation de la conception/du produit ≠ la validation du processus. Les organismes de réglementation exigent individuellement la validation de la conception/du produit et la validation du processus, de sorte que ces deux éléments doivent être pris en compte de manière égale lors de la soumission réglementaire.
À quel moment du processus de développement doit-on penser à la validation ? Une entreprise de dispositifs médicaux doit comprendre qu'il n'est jamais trop tôt pour commencer le travail de validation. Une entreprise doit commencer à valider plus tôt que tard pour s'assurer qu'elle est sur la bonne voie et qu'elle résout le bon problème.
La validation (également V&V) étant un processus itératif, elle consomme un bon investissement, si elle est mal planifiée. Une stratégie de test bien définie peut vous aider à optimiser les coûts ainsi que la période de test afin que le produit soit prêt à être commercialisé à temps.
La complexité de toute stratégie de test dépend des technologies à utiliser et des marchés géographiques cibles. La stratégie de test doit couvrir au moins six paramètres mentionnés ci-dessous :
- Géographies ciblées et normes associées ;
- Délai de mise sur le marché ;
- Une norme à suivre avec une version ;
- Laboratoires d'essai - laboratoires internes ou indépendants ;
- Définir la séquence des tests ;
- Présentation du résultat du test
En conséquence, les tests utilisés pour le processus de vérification et de validation doivent également être validés. Cela permet de s'assurer que l'on mesure ce que l'on doit mesurer, car un test erroné produira des résultats erronés en termes de convivialité et de fonctionnalité. Les entreprises de dispositifs médicaux ont besoin d'un processus de V&V efficace et bien documenté, conforme aux réglementations en vigueur.
Conception de dispositifs médicaux : Gestion des risques
Stratégie de migration des risques vs. plan de gestion des risques
Les procédures de gestion des risques pour les dispositifs médicaux sont appliquées dans le cadre d'une norme de conformité internationalement reconnue. ISO 14971:2007 Dispositifs médicaux - "Application de la gestion des risques aux dispositifs médicaux". En outre, les politiques de gestion des risques doivent être intégrées à toutes les étapes de la conception et du développement des dispositifs médicaux et doivent également être associées aux aspects de contrôle de la conception. [10].
La gestion des risques ne s'arrête jamais (du moins en théorie !). La philosophie de la gestion du risque est qu'elle ne doit pas être un ensemble de règles strictes et rapides. La gestion des risques et la stratégie de migration des risques consistent à comprendre l'intention de la gestion des risques et à aborder le processus de manière logique et systématique. En d'autres termes, ne vous contentez pas de suivre les règles... réfléchissez !
Considering the complexity of medical device design, focused risk management practices help ensure usability, safety, and conformité réglementaire. It is a process of identifying, controlling, and preventing the failure that may cause hazards to users. It also mandates identifying associated risks.
La figure 3 montre toutes les étapes du processus de gestion des risques. Le processus commence par l'identification des dangers, puis le risque associé est mesuré sur la base des conséquences des dangers et de leur possibilité de risque.
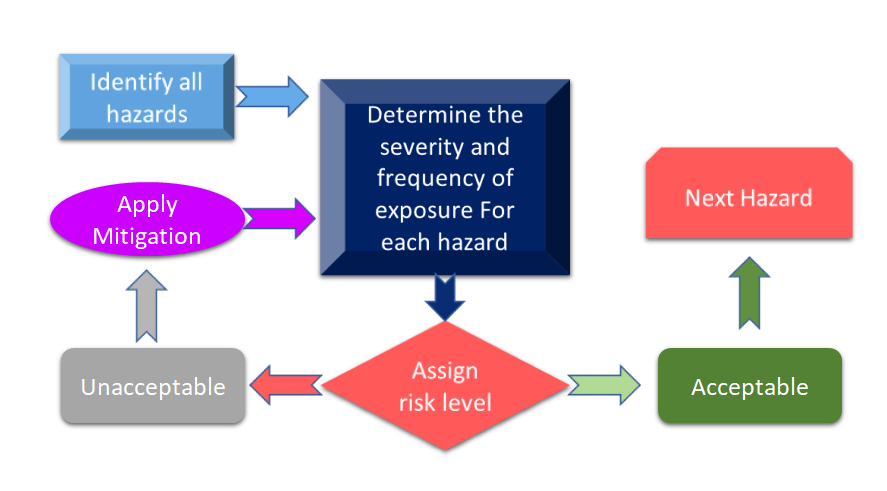
Figure 3 : Processus de gestion des risques pour les dispositifs médicaux (adapté de [11]).
Si le risque identifié au cours du processus de gestion des risques pour les dispositifs médicaux est supérieur aux critères définis, il vous faudra alors procéder à une atténuation des risques. Le niveau de risque dépend de plusieurs paramètres, notamment de votre dispositif, des technologies et, dans certains cas, de la manière dont votre entreprise gère le processus d'atténuation des risques. Il est toujours conseillé d'effectuer une analyse des risques pour votre dispositif afin de voir quelles normes peuvent être appliquées à votre dispositif. Dans la récente révision de la norme ISO 14971 : Norme internationale pour la gestion du risque des dispositifs médicaux, l'analyse du risque et l'analyse préliminaire des dangers (PHA) sont identifiées comme des exigences primordiales pour votre dispositif médical. [12]. D'une manière simplifiée, l'ASP est censée fournir le cadre initial pour l'évaluation et la gestion des risques et l'ASP couvre à la fois l'analyse et l'évaluation des risques. Selon la définition, l'évaluation des risques comporte une liste de dangers, de dommages, de situations dangereuses, formulés à partir des matériaux de construction de vos appareils, des composants ou des matières premières utilisés dans vos appareils, des interfaces homme-appareil ou manuel, de l'environnement d'utilisation, du principe de fonctionnement et d'autres facteurs pertinents. [13].
Conclusion
En fin de compte, pour toute entreprise de dispositifs médicaux en démarrage ou pour une organisation bien établie, il est important de se rappeler que la lecture des règlements ne vous apporte rien, mais que la compréhension de la philosophie vous apporte beaucoup !
En résumé, lorsqu'il s'agit d'analyser et de planifier les risques :
- Doit être utilisé dès le début et tout au long du processus de conception et de développement,
- Génère souvent de nouvelles informations à intégrer dans le processus de conception et de développement (de dispositifs actuels et futurs),
- Aucune planification ne peut éliminer tous les dangers et les risques... mais vous pouvez en atténuer beaucoup ! (Suivre les philosophies de contrôle de la conception décrites ici atténue automatiquement les risques).
Le parcours de chaque dispositif médical jusqu'au marché est complexe en raison des divers facteurs à prendre en considération, tels que les modes d'utilisation, le matériel, l'expérience de l'utilisateur, les réglementations, etc.
Besoin d'aide avec medical device design? Browse experienced experts de l'industrie medtech sur Kolabtree ou postez gratuitement votre projet pour obtenir des propositions.
RÉFÉRENCES ET RESSOURCES
- https://www.welldoc.com/health-plans/
- https://ec.europa.eu/docsroom/documents/10337/attachments/1/translations
- FDA, 2005, Total Product Lifecycle, FDA-CDRH Presentation by CDRH Director Dr. David Feigal, http://www.fda.gov/cdrh/strategic/presentations/ tplc.html
- Pietzsch, Jan & Shluzas, Lauren & Paté-Cornell, Marie-Elisabeth & Yock, Paul & Linehan, John. (2009). Stage-Gate Process for the Development of Medical Devices. Journal of Medical Devices. 3(2).
- Stratégies réglementaires pour la troisième édition de la CEI 60601-1 Consulté le 9 septembre 2020.
- https://www.meddeviceonline.com/doc/an-introduction-to-international-medical-device-standards-0001
- https://www.fda.gov/files/drugs/published/Design-Controls—Devices.pdf
- Feigal DW. Annexe D. Impact du cadre réglementaire sur le développement et l'innovation des dispositifs médicaux. Comité de l'Institute of Medicine (US) sur le Santé publique Effectiveness of the FDA 510(k) Clearance Process; Wizemann T, editor. Public Health Effectiveness of the FDA 510(k) Clearance Process: Balancing Patient Safety and Innovation: Workshop Report. Washington (DC): National Academies Press (US); 2010. Appendix D, Impact of the Regulatory Framework on Medical Device Development and Innovation. Available from: https://www.ncbi.nlm.nih.gov/books/NBK209794/.
- 1997, FDA CDRH 1997, Design Control Guidance for Medical Device Manufacturers (Guide de contrôle de la conception pour les fabricants de dispositifs médicaux).
- https://starfishmedical.com/blog/iso-134852016-section-7/?doing_wp_cron=1599995964.4528369903564453125000
- Teixeira, M. B., et Bradley, R., 2003, Design Controls for the Medical Device Industry, Marcel Dekker, New York.
- ISO 14971:2019 - Dispositifs médicaux - Application de la gestion des risques aux dispositifs médicaux
- ISO/TR 24971:2020 - Dispositifs médicaux - Guide d'application de l'ISO 14971
Tous les articles de cette série :
Développement et conception de dispositifs médicaux : Un guide définitif
Développement de dispositifs médicaux : 3 conseils pour réussir
Medical Device Design : Le guide essentiel, étape par étape
Commercialisation des dispositifs médicaux : 9 étapes de l'esquisse au lancement
Comment surmonter les défis de la commercialisation des dispositifs médicaux
Lancement de dispositifs médicaux : Étapes clés pour mettre votre produit sur le marché
Surveillance post-commercialisation des dispositifs médicaux : Un guide complet
Kolabtree helps businesses worldwide hire freelance scientists and industry experts on demand. Our freelancers have helped companies publish research papers, develop products, analyze data, and more. It only takes a minute to tell us what you need done and get quotes from experts for free.

Unlock Corporate Benefits
• Secure Payment Assistance
• Onboarding Support
• Dedicated Account Manager
Sign up with your professional email to avail special advances offered against purchase orders, seamless multi-channel payments, and extended support for agreements.


