



Biólogo computacional y Kolabtree freelance Shaurya Jauhari writes about current challenges involved in conducting pathway analysis in bioinformatics and possible solutions to the problem.
In the contrasting era of “Imprecise Medicine” (propelling us to tune towards Precision Medicine) and inundation of biomedical data brought forth by advancements in instrumentation technologies, a lacuna persists that is largely premised over mapping data to information. The clinical experiments engender biomarcadores (technically list of genes or genomic regions more contemporarily) that have to be expounded for their biological implications. The current suite of tools that facilitate such an endeavor is less purposeful as it neglects the ipso facto, las interacciones espaciales del genoma dado su perfil de acomodación dentro del núcleo de cada célula eucariota. Este comentario trata de poner de relieve la naturaleza del problema, arrojar luz sobre la organización del genoma, reflexionar brevemente sobre las herramientas actuales de cartografía y conjeturar las posibles soluciones.
El diablo está en los detalles
Un grupo de esfuerzos para perfeccionar la resolución de los datos genómicos está rozando un esquema crucial de información. Estamos impulsando ansiosamente la probabilidad de un genoma $1000, aunque nos importa menos el análisis $100.000. Existe un gran compendio de repertorios que contienen las anotaciones a los resultados experimentales y los casos bajo un estudio biológico típico. Puede haber definiciones que indiquen las implicaciones biológicas de que un gen, o la vía de la que forman parte esos genes, se asocie a una enfermedad. Una vez más, este tipo de almacenes dinámicos de información han sido curados manualmente (antiguamente) y la gestión del conocimiento ha sido asumida por tuberías automatizadas que emplean ordenadores y TIC en general. Estas bases de datos se actualizan con una sabiduría científica orientada al consenso y han acogido un puñado de revisiones desde su creación. El conducto que relaciona los resultados experimentales con sus implicaciones biológicas está muy atenuado, en gran medida porque se descarta la "verdadera" biología subyacente.
Nuestro genoma, con una media de 2 metros de longitud, se aloja en el núcleo de cada una de las células que conforman nuestro cuerpo. Debido a la diminuta estructura de una célula y, sobre todo, de su núcleo, el genoma está empaquetado de forma un tanto forzada y blanda. Esto permite que regiones del genoma, bastante distantes desde una perspectiva lineal, se acerquen e interactúen. Este adagio es rechazado por el actual conjunto de herramientas de enriquecimiento (mapeo) y, por lo tanto, los resultados generados son desproporcionados.
Las regiones del genoma forman parte de grandes "grupos de acción" o vías de acceso that are technically series of chemical reactions accounting for a phenotype; healthy or diseased. When a diseased state is examined, the investigators are on the lookout for the biomarkers that have potentially gone awry and have apparently transformed the organismal body from tonificado a se movió. Imagínese persiguiendo una enfermedad difícil de combatir con información desajustada.
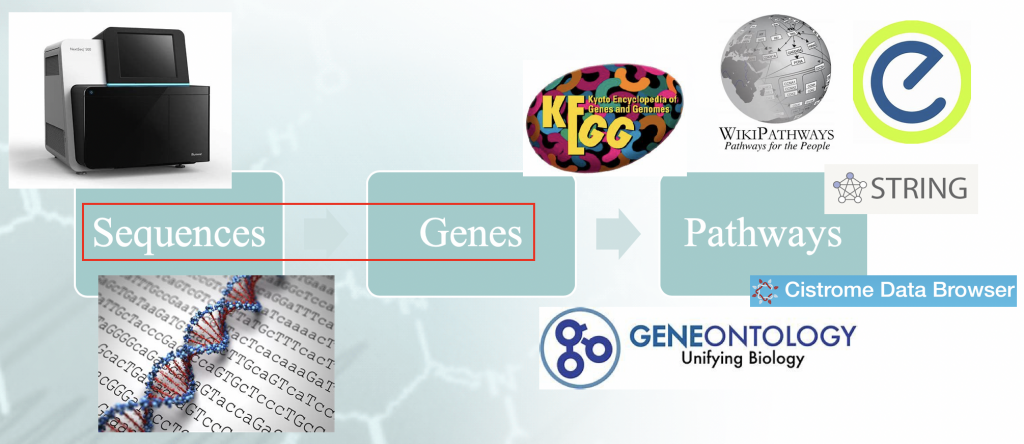 Planteamiento del problema - Flujo de trabajo del típico análisis de enriquecimiento. Hay una cierta
Planteamiento del problema - Flujo de trabajo del típico análisis de enriquecimiento. Hay una cierta
"idiomatismo" asociado al mapeo de las secuencias del genoma a los genes, y que orquesta la
resultados de la corriente.
Organización del genoma
Como ya se ha dicho, el genoma es lo suficientemente largo como para ser almacenado linealmente dentro del núcleo de una célula, para cada célula de nuestro cuerpo o de cualquier otro organismo vivo. Más bien, esta entidad de 2 metros está aplastada y apiñada en una estructura aparentemente desordenada, con bucles, giros y remolinos, como uno puede imaginar. Este interminable La cadena de nucleótidos o pares de bases -Adenina (A), Citosina (C), Timina (T) y Guanina (G)- estructura distintas topologías dentro del núcleo mientras se ajusta a la exigüidad. Forman bucles de cromatina, compartimentos/subcompartimentos, dominios/subdominios que cumplen una función, de acuerdo con el tipo de célula. (Obsérvese que los distintos tipos de células funcionan de manera diferente; una célula nerviosa tiene otras empresas que atender que una célula muscular; cada una tiene un papel exclusivo que desempeñar).
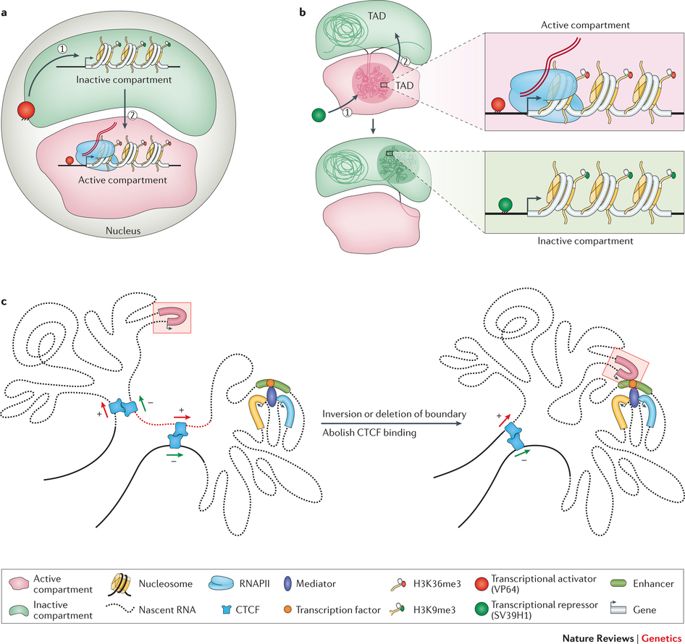
El genoma se enrolla y se extiende en espacios objetivos. (Crédito: https://doi.org/10.1038/nrg.
2016.112)
Bases de datos de rutas
Existen diversas ontologías y bases de datos, entre ellas la Enciclopedia de Genes y Genomas de Kioto (KEGG) (https://www.genome.jp/kegg/) y Gene Ontology (http://geneontology.org/Las pocas herramientas que voy a exponer en la próxima sección (y que a menudo son objeto de opinión) engendran términos de enriquecimiento que proceden "selectivamente" de dichas bases de datos. A partir de sus valores de significación estadística, se deduce si realmente representan un fenotipo de la lista o son sólo una cuestión de desarrollo aleatorio. (P.D. Hay un escrito sobre los valores p que presumiblemente ayudará a los profanos a entender la idea de significación estadística. Por favor, siga el enlace https://sway.office.com/WkyHrPnVB8Ec3zPD?play a su conveniencia).
Herramientas de enriquecimiento
El análisis de enriquecimiento es un protocolo computacional de novo regiones genómicas a sus definiciones registradas en las bases de datos a las que se ha aludido. Por lo que respecta a las herramientas (que actúan como conducto), se estructuran clásicamente bajo varios epígrafes, a saber, análisis de sobrerrepresentación (ORA), puntuación de clases funcionales (FCS), métodos basados en la topología de las vías (PT) y métodos de interacción de redes (NI).
Análisis de la sobrerrepresentación
El análisis de sobrerrepresentación, a través del dogma de la distribución hipergeométrica, evalúa el conjunto de genes diferencialmente expresados para los que podrían formar parte de una vía biológica. Básicamente, una prueba hipergeométrica considera cuatro atributos para llegar a una decisión, a saber
- Número total de genes en el ensayo considerado,
- Los genes expresados diferencialmente,
- Genes en la vía objetivo del número total de genes, y
- Genes expresados diferencialmente en la vía objetivo.
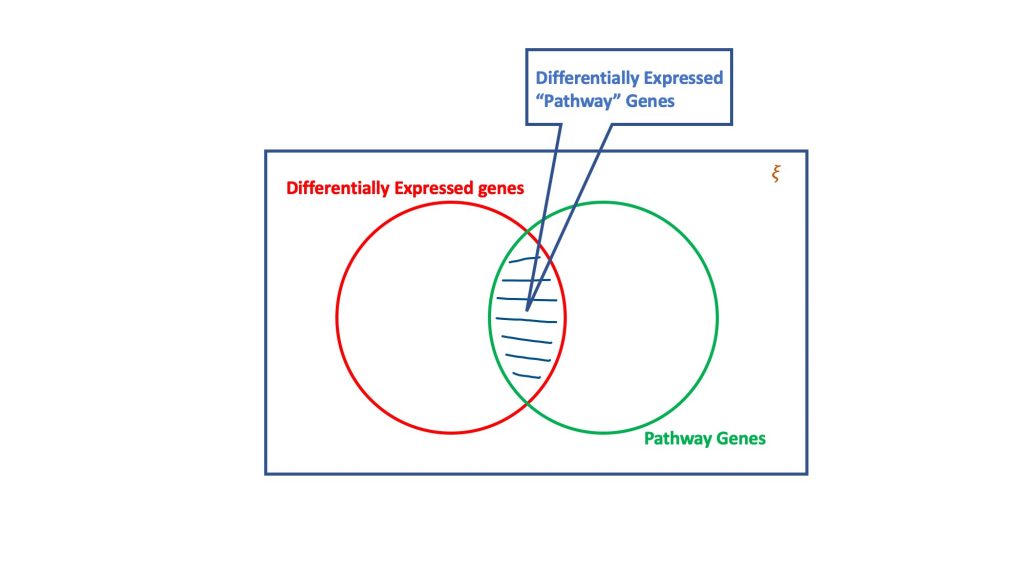
La esencia de la prueba hipergeométrica
Despite being a simple and straightforward methodology, ORA presents its own limitations.
- Democracia en juego; todos los genes se consideran por igual¿Por qué es un problema? Supongamos que los genes se filtran en función de cambio de pliegues. Seleccionamos los genes que abarcaban una diferencia de expresión mayor o igual a 2 veces (pliegues), tanto en sentido negativo como positivo. Aunque el mínimo era de 2 veces, este flujo de trabajo también capturaría genes con cambios de expresión de 3 veces, 4 veces y más. Seguramente, un gen con una disparidad de expresión de 4 veces es más prudente que un gen con un cambio de 2 veces. Esta manifestación está descartada por la ORA.
- Considera sólo los genes más significativosDe nuevo, consideremos un gen con un cambio de pliegue de 1,9999 o un valor p <0,0051113; normalmente, un valor p <0,05 se considera estadísticamente significativo. La metodología ORA pasa por alto este gen en el resultado final. Está claro que hay una pérdida de información y una falta de flexibilidad. (P.S. Breitling et al. abordaron este problema proponiendo una extemporaneidad para evitar los umbrales. La revisión emplea un enfoque iterativo que añade un gen a la vez para compilar un conjunto de genes para los que una vía es óptimamente significativa).
- Ningún gen funciona de forma aisladaEsto se debe a las limitaciones mencionadas anteriormente, ya que al tratar el gen como una entidad independiente se pierde la esencia de la contribución poligénica a un fenotipo. Uno de los objetivos del análisis de la expresión génica podría ser dilucidar cohortes de genes cuyos patrones de expresión sean congruentes. Esta sinfonía pone de relieve los genes funcionalmente afines o los que trabajan hacia un estado biológico común.
- Vías independientes entre síLa ORA también asume que las vías no funcionan en tándem (o en sucesión). Esto es principalmente erróneo, ya que una serie de reacciones químicas podría preceder o proseguir una a otra.
Puntuación de la clase funcional
Contrary to ORA, FCS methods subsume all the contextual genes as well as their association estadísticas (fold-change, p-value) and compute a corriendo Puntuación de enriquecimiento para agrupaciones de genes (basada en algún conocimiento funcional como la Ontología de Genes o las vías KEGG). Por ejemplo, GSEA del Instituto Broad (http://software.broadinstitute.org/gsea/index.jsp). Una ejecución típica de FCS analizaría el cambio de expresión de los genes en general en la lista (no la clasificación por significación estadística o algo más) de genes expresados diferencialmente en un experimento. El resultado principal del análisis de enriquecimiento de conjuntos de genes es una puntuación de enriquecimiento (ES) que refleja el grado de sobrerrepresentación de un conjunto de genes en la parte superior o inferior de una lista clasificada de genes; ¿por qué superior e inferior? porque ahí están los genes más alejados de lo normal, en términos de cambio de expresión. Una puntuación ES positiva para un conjunto de genes (o una vía objetivo, GS) será indicativo de los genes de la lista (GL) que caen en la parte superior (más regulados; 1,2,3 ...), mientras que una puntuación ES negativa significará que los genes componentes se encuentran en la parte inferior (más regulados; n-3, n-2, n-1, n, donde n es el número total de genes). P.D. El ES se convierte en ES normalizado (NES) cuando se corrige el problema de las pruebas múltiples (tasa de falsos descubrimientos, p. ej. Método de Bonferroni).
En resumen, los métodos FCS son notablemente mejores que los métodos ORA por,
- evitando el requisito de un umbral arbitrario para clasificar los genes como significativos o no significativos.
- Apreciar la información sobre la expresión de los genes para rastrear los cambios sistemáticos en la vía; esto permite rendir cuentas sobre la interdependencia de los genes.
Sin embargo, los métodos del FCS también tienen ciertas deficiencias.
- Dado que las vías se analizan de forma independiente, es posible que no se cuenten los genes que regulan varias vías.
- Muchos métodos de SCA clasifican los genes de una lista sobre la base de los cambios en la expresión génica. Un escenario en el que la diferencia de rangos refleja una varianza desigual (y posiblemente exponencial) en la expresión podría ser quizás una medida injusta.
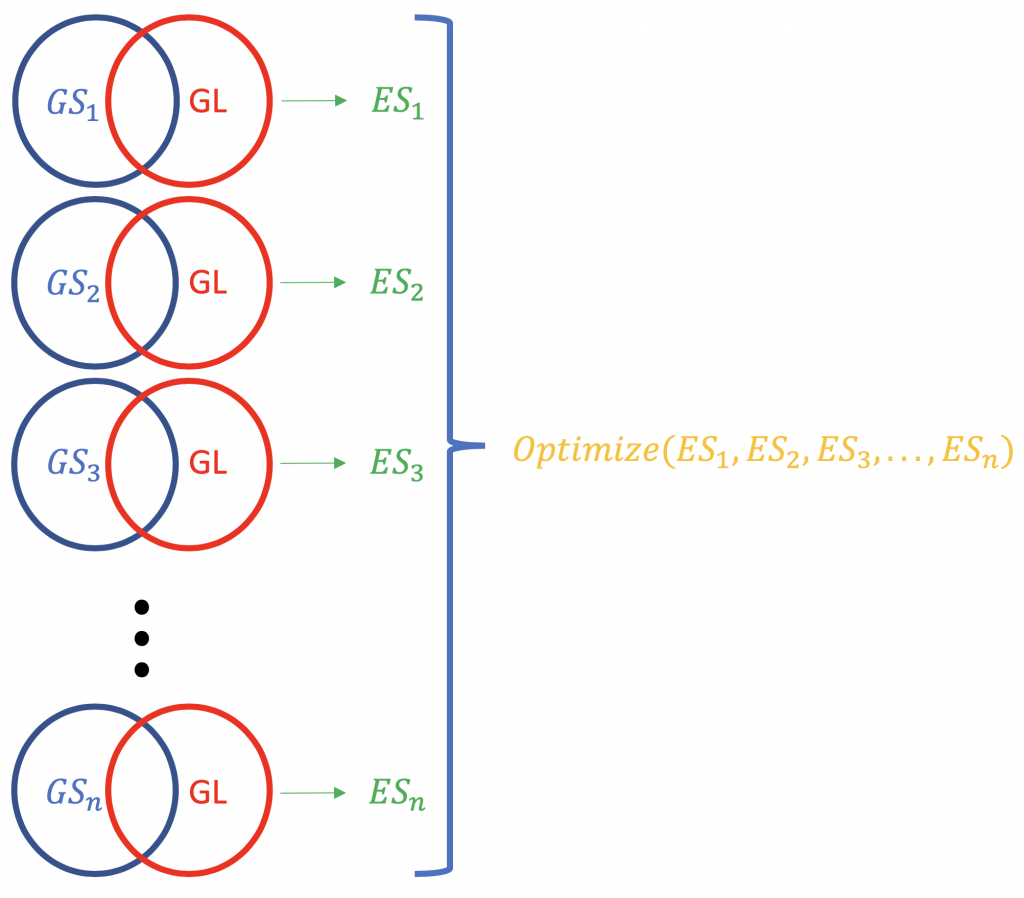
La normalización de las puntuaciones ES correspondientes a una lista de genes.
Enfoques basados en la topología de las vías
Una privación crucial de los métodos ORA y FCS es que ignoran la estructura de las vías. El orden de los genes que se regulan en una vía es esencial para rastrear los efectos causales. Comprensiblemente, podría haber exactamente dos vías con los mismos componentes génicos, pero la jerarquía de activación podría ser totalmente diferente. Si fuera por los métodos ORA/FCS, habrían dado lugar a términos de enriquecimiento similares. Eso es un problema. Los métodos Pathway Topology(PT) asumen la exclusividad en la función dependiendo de las interacciones específicas, lo que se ajusta a la lógica general también. Ejemplos de herramientas son SPIA (https://www.bioconductor.org/packages/release/bioc/manuals/SPIA/man/SPIA.pdf), GGEA y PARADIGM. En general, las herramientas de esta categoría tendrán una puntuación local y otra global. La puntuación local a nivel del gen calibrará los cambios de pliegue en la expresión del gen y de los genes anteriores, mientras que la puntuación global medirá el nivel de la vía para la relación con el conjunto de genes. Sin embargo, esto también hace que los métodos de PT sobreajusten los datos para una condición/tipo de célula peculiar.
Análisis basado en la interacción de redes
Se trata de una categoría bastante infravalorada que todavía se aplica escasamente, a pesar de su antigua formulación. Métodos como EnrichNet , NetPEA (http://www.dx.doi.org/10.1109/BIBM.2013.6732493) have been proposed close to a decade ago, but they haven’t gained much traction because of limited tools available. This facade constraints the dexterity of the theme as no improvements are documented. That turns out to be an open-ended investigación problem.
El aparente Panacea
Probablemente ahora tenga una idea sobre el enriquecimiento fundamental/análisis de vías y el tipo de herramientas que lo ayudan. Sin embargo, como he mencionado antes, todas las herramientas disponibles (que caen en las categorías marcadas) se basan en un parámetro sesgado de una ventana lineal a través de la región consultada. Si los segmentos del genoma que intervienen entran en este marco, aparecen como enriquecidoo no. El verdadero negocio es tener una herramienta que, tal vez, introduzca un centro y diámetro de un círculo hipotético, por así decirlo para destacar la organización 3D basada en las interacciones del genoma para la región.

GREAT ofrece varias opciones de avance para especificar la dimensión lineal alrededor del Sitio de Inicio de Transcripción de un gen.
 Enrichr presenta una selección directa del tipo de genoma y del número de genes en la supuesta región lineal.
Enrichr presenta una selección directa del tipo de genoma y del número de genes en la supuesta región lineal.
Como se deduce de la Figura 4, no se aprecia la "verdadera" organización espacial del genoma. Este es un problema y una profunda falla que persiste en el dominio actual del análisis de enriquecimiento. A pesar de ello, también hay una relevancia evidente de fábricas de transcripciónidentificados como sitios en el espacio nuclear que atraen a los elementos reguladores distantes para "festejar en casa". En broma, suelo comentar que, al igual que alguien que está enfadado con alguien o algo, las fábricas de transcripción (personificadas) podrían reprender al genoma.La transcripción se hará sobre mi cadáver, ¡nada más!". El tema adyacente de las fábricas de transcripción es objeto de un debate adyacente y futuro. Sin embargo, materializa el dogma de las interacciones cis-reguladoras que está mal en la práctica contemporánea.
Para terminar, me gustaría señalar que el análisis de las vías es una fracción crucial, y a menudo descuidada, de la Bioinformática de la tubería. Siempre hay margen para ampliar las metodologías existentes en función de los datos genómicos, que evolucionan mientras hablamos. Cuando dispongamos de un mayor volumen de datos, no sólo será un problema de infraestructura, sino también de algoritmos.
Necesidad de contratar a un consultor en bioinformática? Trabaja con científicos autónomos en Kolabtree. Es gratis publicar tu proyecto y obtener presupuestos de expertos.
Expertos relacionados:
Autónomo de bioinformática | Genética vegetal | Biología del desarrollo | Terapia génica | Células madre |
Análisis de datos de secuenciación de ADN |Genética animal | Interacciones con otros medicamentos | Genética y genómica
Kolabtree helps businesses worldwide hire freelance scientists and industry experts on demand. Our freelancers have helped companies publish research papers, develop products, analyze data, and more. It only takes a minute to tell us what you need done and get quotes from experts for free.

Unlock Corporate Benefits
• Secure Payment Assistance
• Onboarding Support
• Dedicated Account Manager
Sign up with your professional email to avail special advances offered against purchase orders, seamless multi-channel payments, and extended support for agreements.


