



Submissões da FDA consultor e redator regulador Samradni Patil fornece um Lista de verificação das submissões 510k to help dispositivo médico empresas com autorização rápida e fácil da FDA.
O processo de submissão 510(k) é usado tipicamente para dispositivos médicos Classe II a fim de obter liberação da US Food and Drug Administration (FDA). O processo de aprovação prévia ao mercado (PMA) é normalmente usado para dispositivos médicos da Classe III.
O processo de revisão 510(k) determina a Equivalência Substancial (SE) com um dispositivo similar legalmente comercializado, também chamado de dispositivo predicado. O dispositivo precisa ser pelo menos tão seguro e eficaz quanto o dispositivo legalmente comercializado para afirmar que é substancialmente equivalente a ele. O dispositivo com menos de 510(k) de revisão precisa mostrar o seguinte para reivindicar o SE com o dispositivo predicado:
- O mesmo uso previsto que o dispositivo legalmente comercializado (dispositivo predicado) predicado
- As mesmas características tecnológicas do dispositivo predicado ou
- Diferentes características tecnológicas e informações/teste sugerem que o dispositivo é tão seguro e eficaz quanto o dispositivo predicado e diferentes questões sobre segurança e eficácia do que o dispositivo predicado não são levantadas.
O não cumprimento dos critérios acima leva à determinação da Equivalência Não-Substancial (NSE).
510k Lista de Verificação de Submissões
O processo de revisão do FDA 510(k) pode ser amplamente dividido em 2 etapas.
- Revisão de Aceitação
- Revisão Substantiva
Cronogramas de revisão
| Tipo de revisão | Linha do tempo (Dias do calendário) | Resultado do processo |
| Revisão de Aceitação | No dia 15 | O FDA informa ao solicitante se o pedido é aceito para Revisão substantiva ou Colocado no suporte do RTA |
| Revisão Substantiva | No dia 60 | Revisão interativa ou Pedido de informações adicionais |
Nota: O dia 1 é o dia em que a FDA recebe a aplicação 510(k)
Vamos primeiro discutir que tipo de questões são enfrentadas pelas empresas de produtos médicos durante estes processos de revisão.
Revisão de Aceitação
Se o 510(k) não for aceito nesta fase, ele é colocado em Recusar-se a aceitar (RTA) Manter. De acordo com a Dados da FDAem 2018, aproximadamente 30% 510(k)s foram colocados em um porão da RTA.
Revisão Substantiva
O gráfico abaixo mostra a porcentagem de solicitações de informações adicionais emitidas pela FDA durante a fase de revisão substantiva.
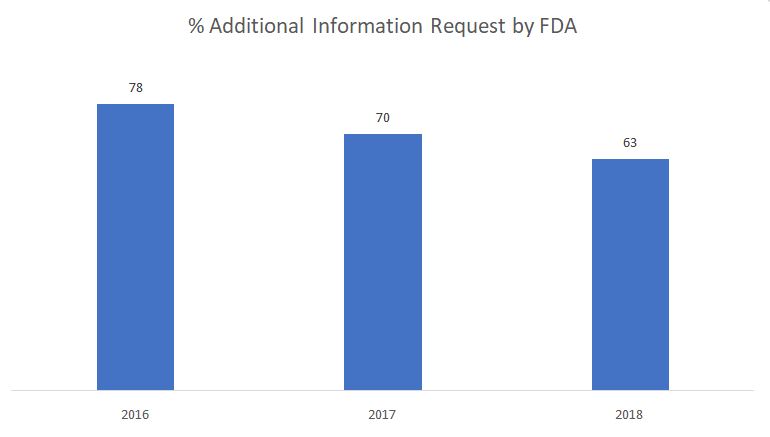
Fonte: FDA
A porcentagem de 510ks determinada como Não-Substancialmente Equivalente (NSE) é mostrado abaixo.
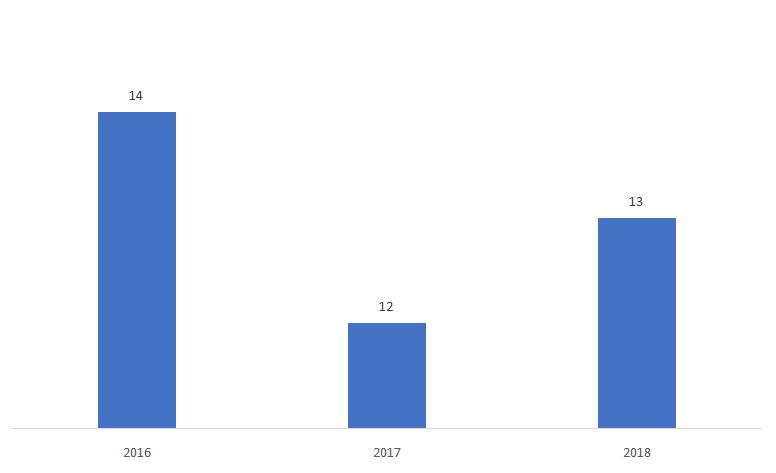
Fonte: FDA
Pedidos de informações adicionais comuns de lista de verificação de envio 510k
Agora que entendemos as questões comuns levantadas como parte do processo de revisão da 510k, vamos nos concentrar em que tipo de pedidos de informações adicionais são comuns.
O Dados da FDA descreve os seguintes tipos de pedidos de informações adicionais:
- Descrição inadequada do dispositivo
- Discrepâncias durante a apresentação - As discrepâncias nesta categoria estão mais freqüentemente relacionadas à descrição do dispositivo ou às indicações de uso
- Problemas com indicações de uso
- Falha em seguir ou abordar o(s) documento(s) de orientação ou normas reconhecidas
- Faltam completamente os testes de desempenho necessários para certos tipos de dispositivos (ou seja, não são fornecidos quaisquer dados de desempenho)
- Faltam completamente os dados clínicos necessários para certos tipos de dispositivos (ou seja, não são fornecidos quaisquer dados clínicos)
Agora, vamos discutir as melhores práticas a seguir durante a preparação e apresentação de um pedido de 510k.
510k lista de verificação de envio:
1. Recusar-se a aceitar (RTA) carta
O objetivo da revisão de aceitação no estágio inicial é verificar se o pedido 510k está administrativamente completo. É altamente recomendável rever a lista de verificação de aceitação fornecida no documento de orientação".Recusar-se a aceitar a política para 510ks”.
Para garantir uma revisão de aceitação bem sucedida, sugere-se que cada empresa siga os elementos da tabela a seguir do documento de orientação.
- Tabela de Perguntas Preliminares: Embora esta lista de verificação se destine ao revisor líder a fazer a determinação inicial, é altamente recomendável responder informalmente a estas perguntas antes de apresentar o pedido à FDA.
- Tabela de Elementos Organizacionais: Estes elementos ajudam a organizar o 510(k) de forma a permitir a fácil identificação das informações na aplicação 510(k).
- Tabela de Elementos de uma Apresentação Completa (Itens RTA): As empresas devem prestar mais atenção aos elementos listados nesta tabela, pois estes elementos são críticos para não receber uma carta de RTA.
2. Descrição do Dispositivo Inadequado
A descrição do dispositivo é obrigatória na aplicação 510(k). Sugere-se acrescentar uma breve descrição e especificações técnicas nesta seção. Todos os modelos de dispositivos médicos e acessórios devem ser incluídos. Fotos, diagramas, dimensões, desenhos e tolerâncias para cada componente devem ser incluídos. A falta de um modelo ou acessório importante pode levar a confusão e perguntas adicionais. Especificações técnicas inapropriadas podem levar a concepção errônea e solicitação de testes adicionais.
3. Informações incoerentes durante todo o processo de envio
- Inconsistência na descrição do dispositivo: Se uma empresa decidir fazer a apresentação 510(k) para adicionar modelo adicional, é importante que as seções aplicáveis como carta de apresentação, descrição do dispositivo, rotulagem, discussão de equivalência substancial, seções relacionadas ao desempenho estejam alinhadas com a mudança real. A incoerência pode levar a atrasos administrativos e, na pior das hipóteses, a solicitação de testes adicionais.
- Inconsistência na indicação de uso: Semelhante à descrição do dispositivo, a inconsistência na indicação para uso em várias seções do 510(k) pode causar problemas. A indicação para a declaração de uso é muito importante para fazer a determinação SE.
Inconsistências em várias seções poderiam ser facilmente evitadas através de uma revisão cuidadosa da apresentação antes de ser submetida à FDA. É sempre uma boa idéia ter um conjunto extra de olhos para dar uma olhada em várias seções a fim de evitar tais erros.
4. Uso pretendido diferente do Dispositivo Predicado
Para obter a determinação SE da FDA, o dispositivo precisa ter o mesmo uso previsto que o dispositivo predicado. Isto é importante porque o uso pretendido diferente do dispositivo predicado pode levar a diferentes problemas de segurança e eficácia. Nesse caso, o 510(k) pode não ser um caminho apropriado para obter a liberação do produto. Deve-se observar que as diferenças na indicação de uso entre o dispositivo predicado e o dispositivo sob a análise do 510(k) podem não resultar necessariamente em um uso pretendido diferente. As empresas devem envidar esforços adicionais para mostrar claramente que as diferenças na indicação de uso não resultaram em uso pretendido diferente.
A orientação da FDA "O Programa 510(k): Avaliando a Equivalência Substancial em Notificações Pré-Mercado [510(k)]"deve ser encaminhada para obter clareza sobre as questões relacionadas ao uso pretendido.
5. Informações inadequadas/faltas nos testes
- Informações de teste inadequadas:
É importante compreender os testes aplicáveis a qualquer dispositivo em particular. Com base em um tipo de teste de segurança elétrica de um dispositivo, testes de compatibilidade eletromagnética (EMC), testes de biocompatibilidade, testes de validação de software, testes de esterilização, testes de usabilidade podem ser necessários para reivindicar a segurança e eficácia de um dispositivo.
Muitas vezes, as empresas subestimam a quantidade de testes necessários ou tentam fornecer razões para não realizar nenhum teste em particular.
Exemplo: As empresas podem confiar em dados de biocompatibilidade de um dispositivo similar para reivindicar biocompatibilidade para seu produto. Esta abordagem pode ser aceitável em alguns casos. Entretanto, muitas vezes o processo de fabricação entre esses dispositivos pode justificar testes de biocompatibilidade separados no dispositivo sob a análise 510(k).
Tais testes adicionais podem levar várias semanas. Se o FDA solicitar a realização destes testes adicionais durante a revisão do 510(k), ele adiciona muito tempo na liberação final do 510(k).
Appropriate teams should give thorough consideration to current FDA guidance, product design, risk management process to make determination about amount of testing required.
- Informações sobre testes ausentes:
É importante entender a diferença entre Tradicional 510(k) e Especial 510(k). Todos os dados de teste devem ser incluídos na apresentação do Traditional 510(k).
6. Falha em Seguir ou Endereço de outra forma Atual Documento(s) de Orientação ou Normas Reconhecidas
Como discutido acima, garantir que os testes sejam conduzidos para demonstrar a conformidade com as mais recentes normas reconhecidas. Pode haver mudanças significativas na última versão da norma em comparação com a versão anterior. Isto pode resultar em perguntas adicionais sobre segurança e eficácia se o dispositivo não for testado com a versão mais recente da norma.
A referência aos últimos documentos de orientação ajuda a compreender as expectativas ou recomendações da FDA. Isto também facilita o processo de revisão da FDA, pois a submissão é redigida no formato de fácil compreensão pelo revisor.
7. Determinação da NSE
O objetivo final do processo 510(k) é determinar a Equivalência Substancial (SE) com um dispositivo predicado. Quando informações adicionais são solicitadas pela FDA durante a fase de revisão substantiva, as empresas devem rever cuidadosamente cada solicitação e fornecer uma resposta cientificamente sólida. O não fornecimento dos dados/respostas solicitadas pode resultar na determinação da NSE. Muitas determinações da NSE são devidas à falta de fornecimento de dados de desempenho.
É altamente recomendável consultar e colaborar com a FDA para entender as expectativas nesta fase. Aqui estão alguns outros erros comuns que eu notei em minha experiência.
Aspectos administrativos
8. Apresentação do pedido no endereço correto
Isto é facilmente evitável por erro humano. Consulte sempre o site da FDA para o endereço correto para apresentar sua solicitação.
9. Incluindo cópia impressa, bem como o eCopy de acordo com a última recomendação da FDA
A FDA tem exigências específicas para o número de cópias impressas e eCopies que precisam ser submetidas para o pedido 510(k). Atualmente,1 cópia impressa e 1 eCopy é necessária. Não faça nenhuma suposição. Consulte o site da FDA antes de tirar qualquer conclusão.
10. questões relacionadas ao eCopy
A apresentação deve atender aos padrões técnicos do eCopy. Consultar a seção Orientação eCopy para preparar o eCopy do aplicativo.
A convenção de nomenclatura em PDF e a recomendação do tamanho do arquivo devem ser seguidas para evitar a carta de retenção do eCopy. Embora o uso do Ferramenta eSubmitter-eCopies é voluntária, esta ferramenta ajuda a validar o eCopy de acordo com as recomendações da FDA.
11. Envio de informações ao revisor
Após receber a carta inicial da FDA (RTA hold, eCopy hold), as empresas freqüentemente cometem um erro ao submeter informações ao revisor. Verifique o e-mail recebido da FDA ou orientação apropriada da FDA sobre para onde enviar a resposta à carta de retenção.
Aspectos técnicos
12. Tradicional 510(k) vs Especial 510(k)
As empresas podem cometer um erro ao classificar a aplicação como Tradicional 510(k) ou Especial 510(k). A maior diferença entre o Tradicional 510(k) e o Especial 510(k) é o tempo necessário para analisar a aplicação pelo FDA. O Special 510(k) leva 30 dias corridos, enquanto o Traditional 510(k) leva 90 dias corridos. Vi várias vezes o FDA solicitar às empresas que convertam o Special 510(k) para o Traditional 510(k). Neste caso, as empresas acabam perdendo muito tempo no processo de conversão.
De acordo com a FDA, um Special 510(k) pode ser apropriado quando:
- A mudança proposta é apresentada pelo fabricante legalmente autorizado a comercializar o dispositivo existente;
- Os dados de desempenho são desnecessários, ou se os dados de desempenho forem necessários, métodos bem estabelecidos estão disponíveis para avaliar a mudança; e
- Todos os dados de desempenho necessários para suportar uma equivalência substancial podem ser revistos em um formato de resumo ou análise de risco.
To avoid such mistake, do thorough pesquisa on the FDA database to identify if similar change was submitted as Special 510(k) or Traditional 510(k). Refer to the documentos de orientação da FDA. Se você ainda estiver em dúvida, obtenha ajuda de consultores reguladores. Use uma abordagem baseada no risco. Se ainda estiver em dúvida, recomenda-se adotar uma abordagem conservadora e apresentar um Traditional 510(k).
Desafios exclusivos
13. Natureza do dispositivo
Alguns dispositivos podem levantar questões únicas devido à natureza única do dispositivo. Certas áreas tecnológicas, tais como Inteligência Artificial (IA) e Ciber-segurança são relativamente novas. A FDA tem colaborado com a indústria para desenvolver documentos de orientação nestas áreas.
Nesses casos, é altamente recomendável uma reunião prévia com a FDA antes da apresentação do 510(k). Consulte a Documento de orientação da FDA se as empresas precisarem de feedback e se reunirem com a FDA antes da apresentação do 510(k).
Conclusão
Estes foram os pontos importantes em uma lista de verificação de submissão 510(k). A recusa em aceitar (RTA) cartas da FDA poderia ser facilmente evitada através de uma revisão cuidadosa da submissão e seguindo os documentos de orientação da FDA.
A solicitação de informações adicionais como parte do processo de revisão substantiva poderia ser reduzida por escrito de forma clara e concisa 510(k). Isto é freqüentemente uma combinação de ciência, arte e experiência. É altamente recomendável a ajuda de pessoas experientes. Consultores em Assuntos Regulatórios por escrito 510(k) para evitar erros dispendiosos, siga esta lista de verificação de apresentação 510(k). Mantenha-se atualizado em termos de regulamentos, normas e documentos de orientação aplicáveis para aumentar as chances de sucesso na submissão.
Necessidade de consultar um Especialista em apresentações da FDA? Trabalhar com escritores de regulamentação experientes, especialistas da indústria de dispositivos médicos e 510k consultores que ajudaram as empresas de dispositivos médicos a preparar documentos regulamentares para uma autorização da FDA bem sucedida.
Kolabtree helps businesses worldwide hire freelance scientists and industry experts on demand. Our freelancers have helped companies publish research papers, develop products, analyze data, and more. It only takes a minute to tell us what you need done and get quotes from experts for free.

Unlock Corporate Benefits
• Secure Payment Assistance
• Onboarding Support
• Dedicated Account Manager
Sign up with your professional email to avail special advances offered against purchase orders, seamless multi-channel payments, and extended support for agreements.


