



ハビエル・キレス・オリエテ博士は、経験豊富な freelance bioinformatics consultant on Kolabtree, provides a comprehensive guide to DNA sequencing データ分析, including tools and software used to read data.
はじめに
遺伝情報のほとんどを担う分子、デオキシリボ核酸(DNA)。 生物の.(ウイルスの中には、リボ核酸(RNA)によって遺伝情報が伝達されるものもある)。 DNA分子の基本単位はヌクレオチド(A、C、G、Tの4文字)である。概念的には DNAシークエンス は、DNA分子を構成するヌクレオチドを読み取るプロセスである(例:「GCAAACCAAT」は10塩基のDNA文字列)。現在のシーケンシング技術では、このようなDNAリードが数百万個生成される を、比較的低コストで合理的に行うことができます。参考までに、ヒトゲノム(ゲノムとは、生物のDNA分子の完全なセットのこと)の配列決定にかかる費用は、以下のように下がりました。 $100バリア と、数日でできるようになっています。を配列する最初の取り組みとは対照的です。 ヒトゲノム10年で完成したこのプロジェクトには、約1兆2千億円の費用がかかりました。
This capability to sequence DNA at high throughput and low cost has enabled the development of a growing number of sequencing-based methods and applications. For example, sequencing entire genomes or their protein-coding regions (two approaches known respectively as whole genome and exome sequencing) in disease and healthy individuals can hint to disease-causing DNA alterations. Also, the sequencing of the RNA that is transcribed from DNA—a technique known as RNA-sequencing—is used to quantify gene activity and how this changes in different conditions (e.g. untreated versus treatment). On the other side, chromosome conformation capture sequencing methods detect interactions between nearby DNA molecules and thus help to determine the spatial distribution of chromosomes within the cell.
これらのアプリケーションやその他のDNAシーケンシングのアプリケーションに共通しているのは、数百万のリード配列からなるギガバイトオーダーのデータセットを生成することです。そのため、ハイスループットシーケンシング(HTS)実験の意味を理解するには、充実したデータ解析機能が必要となります。幸いなことに、ほとんどのHTSデータタイプに対して、専用の計算・統計ツールと比較的標準的な解析ワークフローが存在する。初期の解析ステップのいくつかは、ほとんどのシーケンスデータタイプに共通していますが、より下流の解析は、データの種類や解析の最終目的によって異なります。以下では、HTSデータの解析における基本的なステップについて説明し、一般的なツールを紹介します。
以下のいくつかのセクションでは、ショートリードシーケンシング技術から生成されたデータの解析に焦点を当てています(主に イルミナなど)があり、これらが歴史的にHTS市場を支配してきました。しかし、より長いリードを生成する新しい技術(例えば オックスフォード・ナノポア・テクノロジーズ, PacBio)が急速に普及しています。長時間のシークエンスにはエラー率が高いなどの特徴があるため、この種のデータを解析するための特別なツールが開発されています。
ローリードの品質管理(QC
熱心な分析者はFASTQファイルから分析を開始します。 FASTQフォーマット は、長い間、ショートリードのシーケンスデータを保存するための標準となっていました。要するに、FASTQファイルは、塩基配列と塩基ごとの 何百万ものリードの呼び出し品質を向上させます。ファイルサイズは実際のリード数によって異なりますが、FASTQファイルは一般的に大きく(メガバイトやギガバイトのオーダー)、圧縮されています。なお、FASTQファイルを入力として使用するほとんどのツールは、圧縮されたフォーマットで扱うことができますので、ディスクスペースを節約するためには、解凍しないことをお勧めします。慣例として、ここではFASTQファイルをシーケンスサンプルと同一視します。
FastQC は、生のリードのQCを実行するための最も一般的なツールと思われます。このツールは、視覚的なインターフェースまたはプログラムで実行することができます。コマンドライン環境に慣れていないユーザーにとっては前者の方が便利かもしれませんが、後者は比類のないスケーラビリティと再現性を提供します(数十個のファイルに対してツールを手動で実行することがどれほど面倒でエラーを起こしやすいかを考えてみてください)。いずれにしても、FastQCの主な出力は HTMLファイル reporting key summary statistics about the overall quality of the raw sequencing reads from a given sample. Inspecting tens of FastQC reports one by one is tedious and it complicates the comparison across samples. Therefore, you may want to use MultiQCFastQCからのHTMLレポート(アダプタートリミングやアライメントなど、ダウンストリームで使用される他のツールからのレポートも含む)を1つのレポートにまとめます。.
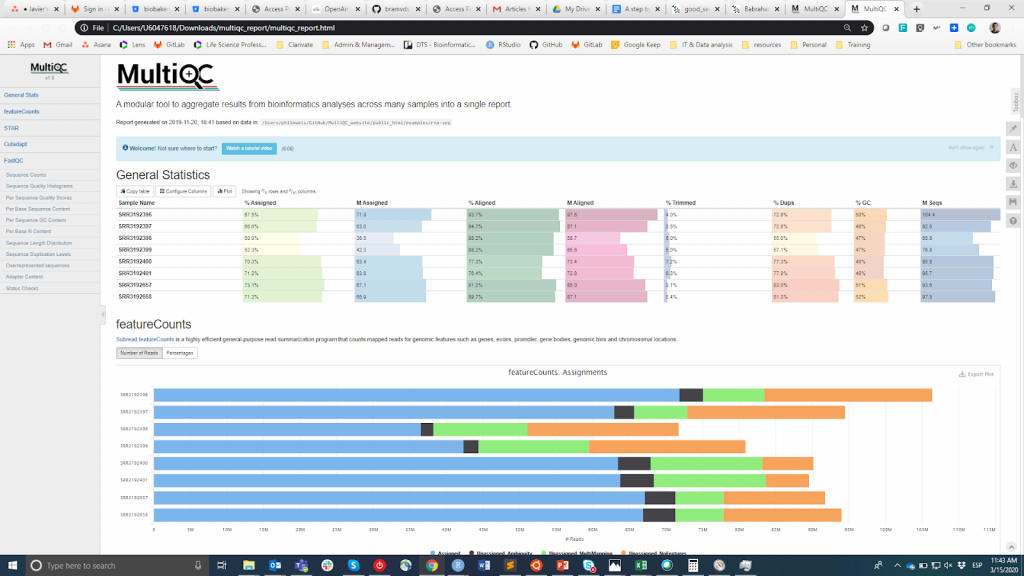
MultiQC
QC情報は、ユーザーがサンプルの品質が良く、後続のステップに使用できるか、廃棄する必要があるかを判断できるようにすることを目的としています。残念ながら、FastQCメトリクスに基づいてサンプルを品質の良いもの、悪いものに分類するためのコンセンサスとなる閾値はありません。私が使用しているアプローチは以下の通りです。同じ手順(DNA抽出、ライブラリ作成など)を経たすべてのサンプルは、品質の統計値が類似しており、大多数の「合格」フラグが立っていると予想します。一部のサンプルの品質が平均よりも低い場合でも、このことを念頭に置いて下流の分析に使用します。一方で、実験中のすべてのサンプルが複数のメトリクスで「警告」または「失敗」フラグを系統的に取得した場合(下記参照 この例)の場合、実験で何か問題が発生したと考えられるので(例:DNAの質が悪い、ライブラリの準備ができていないなど)、再度実験を行うことをお勧めします。
トリミングを読む
生のリードのQCは、問題のあるサンプルを特定するのに役立ちますが、リードの実際の品質を向上させるものではありません。そのためには、リードをトリミングして、技術的な配列や低品質の末端を取り除く必要があります。
技術的な配列とは、実験手順の残り物である(例:配列決定アダプター)。このような配列がリードの真の配列に隣接している場合、アライメント(下記参照)によってリードがゲノムの誤った位置にマッピングされたり、あるアライメントの信頼性が低下したりする可能性があります。また、技術的な配列以外にも、生物学的由来の配列がリードの中に多く含まれている場合には、それらの配列を除去したい場合もあります。例えば、DNAの調製方法が最適でない場合、DNAに変換されたリボソームRNA(rRNA)がサンプル中に高い割合で含まれることがあります。このタイプの核酸がシーケンス実験のターゲットでない限り、rRNAに由来するリードを残しておくと、下流工程の計算負荷が増大し、結果が混乱する可能性があります。なお、テクニカルシーケンス、rRNA、その他のコンタミのレベルが非常に高い場合、おそらくQCですでにハイライトされていると思いますが、その場合は、シーケンスサンプル全体を廃棄した方がよいでしょう。
ショートリードシーケンスでは、DNAの塩基配列を1塩基ずつ(厳密にはシーケンスサイクルごとに1塩基ずつ)決定します。言い換えれば、シークエンスサイクル数がリード長を決定する。HTS法の問題点として、シーケンスサイクルを重ねるごとに、ヌクレオチドを決定する精度が低下することが知られている。これは、塩基ごとのコーリング品質の全体的な低下に反映され、特にリードの最後の方で顕著になります。技術的な配列の場合と同様に、品質の低い末端を含むリードをアラインメントしようとすると、誤配置やマッピング品質の低下につながる可能性があります。
技術的・汚染的な配列や低品質の末端を除去するために、以下のようなリードトリミングツールを使用します。 トリモマティック そして カットダップ が存在し、広く利用されています。このようなツールは、技術的な配列(社内で入手可能なものやユーザーが提供したもの)を取り除き、品質に基づいてリードをトリミングし、リードの長さを最大化します。トリミング後に短すぎるリードは廃棄されます(36ヌクレオチド未満など、過度に短いリードは、ゲノムの複数の部位にマッピングされる可能性が高いため、アライメントのステップが複雑になります)。廃棄されるリードの割合が高いと、データの質が悪いことを示している可能性があるので、トリミングに成功したリードの割合を見てみるとよいでしょう。
最後に、通常はトリミングされたリードに対してFastQCを再実行し、このステップが効果的であり、QCメトリクスが体系的に改善されたことを確認します。
アライメント
例外はありますが(例 デ・ノボ・アセンブリー)、ほとんどのHTSデータタイプとアプリケーションでは、通常、アライメント(マッピングとも呼ばれる)が次のステップになります。リードのアラインメントでは、リードの配列が由来するゲノム上の位置を決定します(通常、染色体:スタートエンドで表されます)。したがって、このステップでは、リードをアライン/マッピングするための参照配列を使用する必要があります。
参照配列の選択は、複数の要素によって決定されます。1つは、配列決定されたDNAの由来となる生物種です。高品質の参照配列が利用可能な種の数は増加していますが、研究が進んでいない生物ではまだそうではない場合もあります。そのような場合には、参照ゲノムが利用可能な進化的に近い種にリードをアラインするとよいでしょう。例えば、コヨーテのゲノムには参照配列がないので、近縁種である犬のゲノムをリードのアライメントに利用することができます。同様に、より高品質な参照配列が存在する近縁種にリードをアラインメントしたい場合もあります。例えば、テナガザルのゲノムは、すでに 公開このような場合、ヒトの参照配列を用いてアライメントを行うことが有効です。
また、配列の更新や改良に伴い、新しいバージョンがリリースされるため、基準配列アセンブリのバージョンも考慮する必要があります。重要なのは、あるアライメントの座標がバージョンによって異なることです。例えば、ヒトゲノムの複数のバージョンは UCSC Genome Browser.いずれにしても、最新のアセンブリバージョンが完全にリリースされたら、そのバージョンに移行することを強くお勧めします。移行中は、既存の結果が古いバージョンとの比較になってしまうため、多少の迷惑がかかるかもしれませんが、長い目で見れば得策です。
また、シーケンシングデータの種類も重要です。DNA-seq、ChIP-seq、またはHi-Cプロトコルから生成されたリードは、ゲノムの参照配列にアライメントされます。一方、DNAから転写されたRNAは、さらにmRNAに加工される(イントロンが除去される)ため、多くのRNA-seqリードは、ゲノム参照配列にアライメントできません。そのため、トランスクリプトーム参照配列にアラインするか、ゲノム配列を参照する場合はスプリットを考慮したアライナー(後述)を使用する必要があります。これに関連して、参照配列のアノテーション、つまり、遺伝子、転写産物、セントロメアなどの座標を持つデータベースのソースの選択があります。私が通常使用するのは GENCODEアノテーション 網羅的な遺伝子アノテーションと転写産物の配列を組み合わせているからです。
ショートリード配列のアライメントツールは数多く開発されています(「ショートリード配列のアライメント」の項参照)。 これ). Reviewing them is beyond the scope of this article (details about the algorithms behind these tools can be found これ).私の経験では、最も人気があるのは ボウタイ2, BWA, HISAT2, Minimap2, スター そして トップハット.私がお勧めするのは、HTSデータの種類などの重要な要素を考慮してアライナーを選択することです。 とアプリケーション、コミュニティに受け入れられているか、ドキュメントの質、ユーザー数などを考慮する必要があります。例えば、RNA-seqをゲノムにマッピングする際には、STARやBowtie2のようにエクソン-エクソン接合部を認識するアライナーが必要です。
多くのマッパーに共通しているのは、実際のアラインメントを行う前に、リファレンスとして使用する配列にインデックスを付ける必要があることです。このステップは時間がかかるかもしれませんが、各参照配列に対して1回だけ行う必要があります。ほとんどのマッパーは、SAM/BAMファイルにアライメントを保存します。 SAM/BAMフォーマット (BAMファイルは、SAMファイルのバイナリ版)。)アライメントは、シーケンスデータの解析において最も計算量が多く、時間がかかるステップであり、SAM/BAMファイルは重い(ギガバイト単位)。したがって、アラインメントを適度な時間で実行し、結果を保存するために必要なリソース(下記の最終セクションを参照)を確保しておくことが重要である。同様に、BAMファイルのサイズとバイナリ形式のため、テキストエディターで開くことは避け、代わりにUnixコマンドまたは以下のような専用ツールを使用してください。 SAMツール.
アライメントから
HTSデータの種類や用途によっても異なるため、アライメント後の明確な共通ステップはないと言えるでしょう。
DNA-seqデータの一般的なダウンストリーム解析は、バリアントコーリングであり、これは、ゲノムの中で、ゲノムリファレンスとの相対的な差異や個人間の差異がある位置を特定することです。このアプリケーションのための一般的な解析フレームワークは GATK 一塩基多型(SNP)や小さな挿入/欠失(インデル)のために(図2).より大きなDNAの塊からなるバリアント(構造的バリアントとも呼ばれる)には、専用の呼び出し方法が必要です(下記参照)。 本記事 を参照してください。)アライナーと同様に、バリアントの種類(SNP、インデル、構造的バリアント)、コミュニティに受け入れられているか、ドキュメントの質、ユーザー数などの重要な要素を考慮して適切なツールを選択することをお勧めします。
RNA-seqの最も一般的なアプリケーションは、遺伝子の発現量の定量化であろう。歴史的には、リードを参照配列にアラインメントし、特定の遺伝子や転写産物にアラインメントされたリードの数を、その発現レベルを定量化するためのプロキシとして使用する必要がありました。このアラインメント+定量化のアプローチは、以下のようなツールによって行われます。 カフリンクス, RSEM または featureCounts.しかし、この手法は、以下のようなソフトウェアに実装された新しい手法に取って代わられつつあります。 カリスト そして サーモン.概念的には、このようなツールでは、リードの全配列を参照配列にアラインする必要はありません。その代わりに、リードがある転写産物に由来すると確信できるだけのヌクレオチドをアラインするだけでよい。簡単に言えば、アラインメント+定量化のアプローチが1つのステップに短縮されるということです。このアプローチは疑似マッピングと呼ばれ、遺伝子発現の定量化のスピードを大幅に向上させます。一方で、疑似マッピングは、フルアライメントが必要なアプリケーション(例:RNA-seqデータからのバリアントコール)には適していないことに注意してください。
シーケンスを用いたアプリケーションで、下流の解析ステップや必要なツールが異なるもう一つの例がChIP-seqです。このような手法で生成されたリードは、ピークコーリングに使用されます。ピークコーリングとは、ターゲットタンパク質が結合していることを示すリードが著しく多い領域をゲノム上で検出することです。いくつかのピークキャラーが存在しており 本誌 を調査しています。最後の例として、Hi-Cデータを紹介します。Hi-Cデータでは、相互作用マトリックスを決定するツールの入力としてアラインメントが使用され、これらからゲノムの3D特徴が決定されます。この記事の範囲を超えたシーケンスベースのアッセイすべてについてコメントします(比較的完全なリストについては 本記事).
始める前に...
この記事の残りの部分では、HTSデータの分析のステップとして厳密には考えられていないかもしれないし、ほとんど無視されている側面に触れています。それに対して、私は、「HTSデータの解析」で提起された質問について考えることが資本であると主張します。 表1 HTSデータ(あるいは他の種類のデータ)の分析を始める前に、私はこれらのトピックについて書いてきました。 これ そして これ.
表1
| 考える | 提案されたアクション |
| 分析に必要なサンプルの情報はすべて揃っていますか? | 実験のメタデータを体系的に収集する |
| 自分のサンプルを明確に識別できるのか? | 各サンプルに固有の識別子を付与するシステムの構築 |
| データや結果はどこにあるのか? | データの構造化・階層化 |
| 複数のサンプルをシームレスに処理することができるのでしょうか? | スケーラビリティ、並列化、自動設定、コードのモジュール化 |
| あなたや他の誰かが、その結果を再現できるでしょうか? | コードや手順を文書化しましょう |
前述のように、HTSの生データとその解析で生成されるファイルの一部はギガバイト単位であるため、数十サンプルを含むプロジェクトではテラバイト単位のストレージが必要になることも珍しくありません。さらに、HTSデータの解析には、計算量の多いステップがある(例:アライメント)。しかし、HTSデータの解析に必要なストレージと計算機のインフラは重要な検討事項であり、しばしば見落とされたり、議論されなかったりする。一例として、最近の分析の一環として、フェノム・ワイド・アソシエーション・アナリシス(PheWAS)を実行している数十の発表論文をレビューしました。最新のPheWASは、100~1,000個の遺伝子変異と表現型の両方を解析するため、重要なデータストレージとコンピューティングパワーを必要とします。しかし、我々がレビューした論文の中には、PheWAS解析に必要なインフラについてコメントしたものはほとんどありませんでした。当然のことながら、私が推奨するのは、直面するであろうストレージとコンピューティングの要件を前もって計画し、コミュニティと共有することです。
DNAシーケンシングデータの解析でお困りですか?ご相談ください。 freelance bioinformatics specialist そして ゲノミクス専門家 をKolabtreeに掲載しました。
Kolabtree helps businesses worldwide hire freelance scientists and industry experts on demand. Our freelancers have helped companies publish research papers, develop products, analyze data, and more. It only takes a minute to tell us what you need done and get quotes from experts for free.

Unlock Corporate Benefits
• Secure Payment Assistance
• Onboarding Support
• Dedicated Account Manager
Sign up with your professional email to avail special advances offered against purchase orders, seamless multi-channel payments, and extended support for agreements.


