



シュリニード・ジョシ, 医療機器エキスパート on Kolabtree, provides a comprehensive guide to 医療機器 design, design controls, validation & verification, regulatory requirements and risk management.
の中にある。 前の記事, we took a look at the overview of the 医療機器 development process from the ideation to the discovery phase. In this article, we will focus more on medical device design, design controls, and compliance.
医療機器設計。IECとISOの規制とコンプライアンス
医療機器を市場に投入するためには、一定の規制要件や規格に適合する必要があることは、すでにご承知のとおりです。国際電気標準会議(IEC)や国際標準化機構(ISO)などの医療機器規格は、医療機器メーカー、設計者、研究所、そしてCDMOなどの医療機器開発サービスプロバイダーが、機器や装置を一定の品質や使いやすさの基準で検査、評価、維持することを可能にしています。
IEC)は、1970年に初の医療機器規格を発表しました。 IEC 60601-1.IEC 60601-1, 医療用電気機器 - Part 1: これは国際的に認められた規格であり,医療用電気機器および装置の一般的な要求事項を規定しており,基本的な安全性と必須性能に関する規格を網羅している。 [4].
IEC 60601-1 は、医療機器分野における最新の医学的発展や技術的進歩に合わせて、定期的に改訂されています。最新の改訂は 2012 年に行われました(IEC 60601-1 の Amendment 1)。この改訂版では、ヒューマンファクターの考慮、医療機器の必須性能評価、ユーザビリティ、コマンドに関する要求事項が含まれています。また、ソフトウェアを医療機器に含め、正式な開発ライフサイクルの採用を規定しています。また、IEC 60601-1 の改訂版の範囲には、ハザード(電気的および機械的)、医療機器のラベリング要件(新しいラベリング規格を含む)、ドキュメントに関する技術仕様の新規および改訂が含まれています。
医療機器のデザイン。ISO規格
また、国際標準化機構には、医療機器の規格の仕様があります。 ISO 13485 そして 国際標準化機構 14971 は、医療機器の品質管理のために世界中で広く使用されている規格です。これらの国際規格以外にも、特定の地域に特化した規格があり、それらはすべて国際規格を若干の修正と制限を加えて採用しています。
医療機器メーカーが米国で医療機器を製造・販売する場合、その医療機器はFDAの規制を受けることになります。ANSI(American National Standards Institute)は、米国におけるISO規格の代表機関です。
さらに似たような組織として、AAMI(Association for the Advancement of Medical Instrumentation)と、アメリカの規格を定めるASQ(American Society for Quality)があります。
医療機器メーカーがISO規格を考慮して機器を設計していた場合、FDAがその機器を承認しない可能性もあります。FDAは、国際規格と地域規格の両方から派生したリスク管理のための独自の手順を持っているため、以下のようなことが考えられます。
(国際規格。)
- ANSI/AAMI/ISO 14971:2007 (R2010), 医療機器 - 医療機器へのリスク管理の適用 (参照した国際規格に追加・修正を加えた地域規格)。) [5].
品質マネジメント規格の場合、国際版や地域版のISO 13485規格には準拠していません。これは、米国市場における医療機器の品質管理について、FDAのガイドラインが異なるためである。
一方、医療機器メーカーが欧州連合(EU)を検討している場合、ISOから採用された規格であるCEN(European Committee for Standardization)と、IECから採用された地域規格であるCENELEC(European Committee for Electrotechnical Standardization)があります。
CENは、ISOからの要求に応じて少し変更され、「EN」という接頭辞を付けて書かれています。例えば、以下のようになります。
各国の会員は、これらの規格をEUから採用し、接頭辞を付けている。スイスの場合、スイス規格は、SN EN ISO 13485:2012 や SN EN ISO 14971:2012 のように、接頭辞として "SN "を付けた規格を発行している。
カナダの場合は、CSA(Canadian Standards Authority)がISOの代表機関となっています。
医療機器の規制とデザインコントロール
医療機器メーカーは、FDA、欧州委員会、カナダ保健省などの規制機関が、医療機器の販売を開始する前に、その医療機器が潜在的なユーザーにとって安全であることを確認したいと考えているため、デザインコントロールのガイドラインに従う必要があります。上のセクションで述べたように、FDAは品質管理に対する要求事項が異なるため、ISO 13485には従っていません。設計管理は以下のように定義されている。 FDA 21 CFR 820.30 これは、ISO 13485 のガイドラインに記載されている 7.3 設計・開発の項と同様の趣旨である。また、FDA は、医療機器の設計における適正品質基準を遵守するために、品質システム規制に cGMP(Current Good Manufacturing Practice)の要求事項を組み込んでいる。 [6].
この規制は、多種多様な機器にデザインコントロールを実施するためのフレームワークを提供します。このフレームワークは、法規制への対応だけでなく、社内の設計・開発プロセスにも柔軟に対応します。
医療機器のデザインコントロールを成功させるためには、経営学、生命科学、工学、コンピュータサイエンス、芸術など、技術的なバックグラウンドと非技術的なバックグラウンドの両方を持つ専門家が必要です。
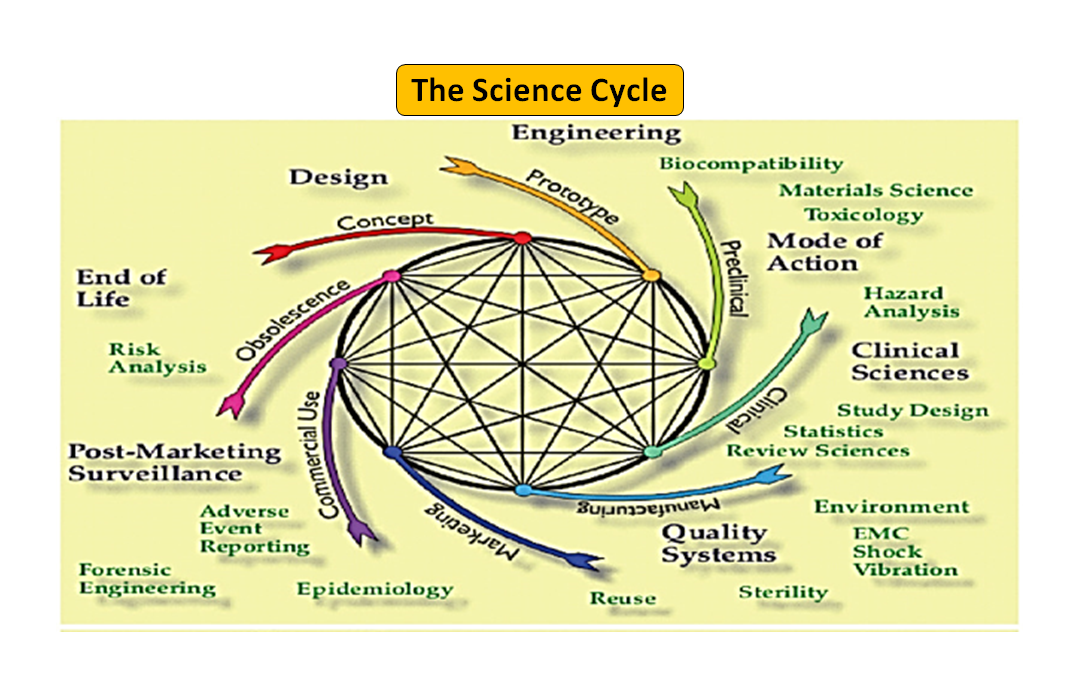
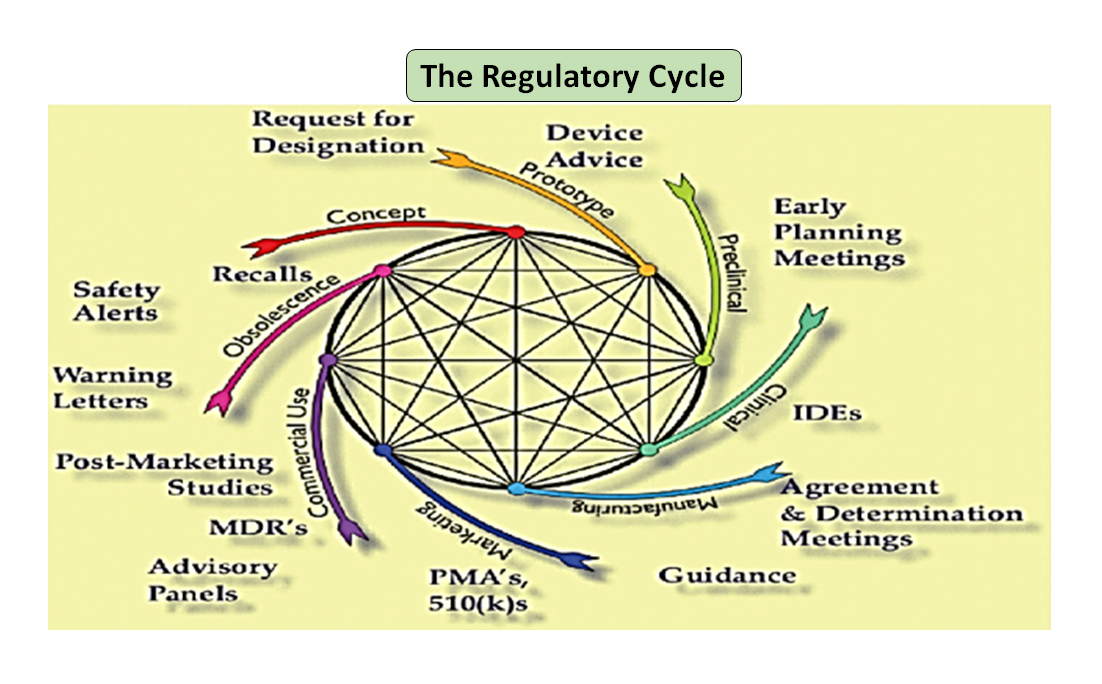
図1: 製品のトータルライフサイクル。サイエンス・サイクルとレギュラトリー・サイクル (Adapted from [7]).
技術革新から薬事承認、商業化までの医療機器のライフサイクルは、機器開発を推進する相互に関連した一連のステップであることは注目に値します(図1:トータルプロダクトサイクルを参照)。初期段階では、エンジニアが設計したプロトタイプをベンチテストして設計を最適化し、生体適合性、抽出物、浸出物、柔軟性、デバイスの全体的な強度などをテストします。薬事アドバイザーの役割は、規制データベースを閲覧して、製品が医療機器として規制されるかどうかを判断するのに役立つガイダンス文書を提案することです。医療機器の使用目的とその動作や作用の様式は、機器設計の指針となり、510(k)、PMA、De Novo、Pre-sub、IDE、HDE、マスターファイルなどの規制経路も決定します。
図1に示すように、科学的プロセスと規制的プロセスの両方が、製品のライフサイクルを通じて相互に絡み合っています。科学のライフサイクルのさまざまな部分が相互に関連しているように、科学と規制要件は相互に関連しており、それぞれが他方に情報を提供し、決定します。ライフサイクルの一部が単独で検討される危険性がないように、FDAとメーカーの両方でつながりを構築する機会があります。例えば、類似製品の市販後の経験を考慮せずに、市販前の申請書が審査されることは珍しくありません。
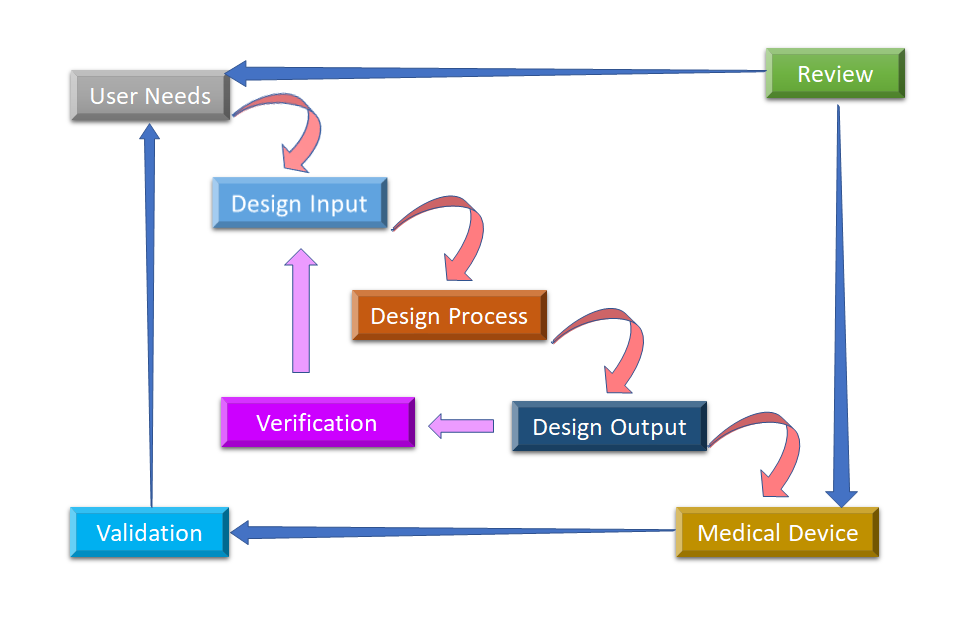
図2: 医療機器のデザインコントロールのためのウォーターフローデザインプロセス(Adapted from [8]).
デザインコントロールが開始される最初の段階は、生産段階で実施されるデバイス設計と製造プロセスからなるDesign Inputの開発と承認です。デザインコントロールは全体的なアプローチであり、デザインが最終的に決定された後、生産段階にデザインを移すだけでは終わりません。設計段階での変更、あるいは製造後のフィードバックに応じて、製造プロセスにも影響を与えます。ユーザーにとって使いやすい製品を開発することは継続的なプロセスであるため、改良された製品では、使用パターンからの革命的な変化や、失敗した製品の分析も考慮されます。図2は、ウォーターフォール型の設計プロセスにおいて、デザインコントロールがどのように行われるかを示したものである。
- ユーザーニーズ:-。 市場のニーズを考慮して要件を定義し、そのニーズを満たすためにデバイスを設計します。進化を重ねた医療機器の設計は、最終的にプロダクションに移管され、製造されます。このプロセスの各段階では、フィードバックが必要となります。
- デザイン入力。 これは反復的なプロセスです。組織が特定のニーズに対応することを決定すると、ニーズから派生した設計入力の受け入れ可能性を検討し、テストします。その時点で、要求を機器設計に変換する反復プロセスが始まります。
- デザインプロセス。 これらの設計インプットは、それらの要求を高レベルの仕様(設計アウトプット)に変換することで、設計アウトプットに変換されます。
- 設計出力。 検証プロセスでは、仕様書が要求を満たしているかどうかを確認します。このプロセスは、設計出力が設計入力と一致するまで続きます。
- 医療機器。 Once the final design is ready, it is transmitted to the production facility for mass manufacturing. Design control regulation mandates Design History File (DHF), which illustrates the linkages and relationships between all the Design Controls and help to trace all changes throughout the entire 製品開発 の処理を行います。
医療機器メーカーでは、紙ベースのアプローチと、デザインコントロール用に開発されたソフトウェアベースのアプローチをとることができます。デザインヒストリーファイルは追跡可能であり、すべてのチームメンバーがアクセスできる必要があります。
下のフローチャートは、医療機器のデザインコントロールのケースを示しています。

医療機器の設計。トレーサビリティーが重要な理由
Currently in the realm of the medical devices industry, it is an ideal practice to develop a traceability matrix that can illustrate the links and relations between user needs, design inputs and outputs, design verification and validation. When you are in the early phase for your device development you can maintain the device traceability using a spreadsheet or document version but as you move forward, its good idea to use cloud-based project management and document sharing platforms such as Microsoft Teams, Asana, Trello or whichever platform is suitable for your organization. The goal is as your project progresses you need to find an option which can save time because the old-school method of maintaining a traceability matrix might consume a lot of your time which you should rather be focusing on design verification and validation.
デザインコントロールのトレーサビリティマトリックスは、製品開発チーム、特にプロジェクトマネージャーにとって不可欠なものである。なぜなら、トレーサビリティはすべてのデザインコントロールの間の関係とつながりを示しているからである。ユーザーニーズはデザインインプットとどのように関連しているか?設計出力は設計入力とどのように関連しているか?設計検証は設計入力と設計出力にどのように関連するか?設計検証はどのようにユーザ・ニーズと関連しているか?トレーサビリティーマトリックスは、医療機器製品の開発の最初から最後までの流れをハイレベルな視点で示す貴重なツールです。
ベストプラクティスの製品開発者は、何年も何年も前からデザインコントロールのトレーサビリティに依存してきました。そして今 ISO 13485:2016 もトレーサビリティを要求しています。ISO 13485:2016 に引用されているように、7.1 製品実現の計画、1.c) 製品に特有の要求される検証、妥当性確認、監視、測定、検査及び試験、取扱い、保管、流通及びトレーサビリティの活動を、製品受け入れの基準とともに、また、7.3.2 設計及び開発の計画、1.e) 設計及び開発のアウトプットから設計及び開発のインプットへのトレーサビリティを確実にする方法。 [9].
医療機器の設計。検証とバリデーション
すべての医療機器が市場で成功を収めるためには、機能性、操作性、信頼性などの目標を満たす必要があります。
さらに、利害関係者(患者、処方者、規制当局、エンドユーザー)は、デバイスの安全性と有効性にも注目するでしょう。例えば、人工呼吸器や心臓病を検出する診断装置など、生命維持に不可欠なアンメットニーズを満たすために設計されたデバイスである可能性が高いです。そのため、検証と妥当性確認を伴うデバイスの反復的なテストは非常に重要です。設計プロセスにおけるこの2つのステップは、医療機器がユーザーの要求に合致しているか、また意図された用途通りに機能しているかを確認することを目的としています。簡単に言えば、設計の検証と妥当性確認は、あなたのデバイスが実際に想定された通りに動作していることを確認することができます。また、設計検証とバリデーションは、医療機器の規制要件、規格、製品品質、製造プロセスを確認することでもあります。設計検証では、設計出力が、設計入力で指定された要件、仕様、または規制要件に準拠しているかどうかを評価します。一方、設計検証は、医療機器がエンドユーザーのニーズに基づいて利益をもたらしているかどうかを評価することを目的としています。
デザイン 検証 を尋ねます。"デバイスの設計は正しかったのか?"
デザイン バリデーション を問いかけます。"Did we design the right device?"
医療機器は、様々な技術の形、サイズ、そして異なるレベルの複雑さで構成されることがあります。検証・妥当性確認(V&V)活動は、規制環境によって推進され、国際標準に従わなければなりません。V&V活動を標準化することで、製造プロセスを合理化し、承認プロセスを向上させることができます。さらに、自動化されたテスト、診断技術、データ収集ツールは、V&Vプロセスを強化することができます。 [10].
- プロダクトバリデーションとプロセスバリデーション
- 医療機器設計/製品バリデーション: - ユーザーや患者のニーズに適合しているかどうか、つまり機器が正しく機能するかどうか。
- プロセスバリデーション:製造プロセスがあらかじめ設定された仕様を満たしていること。
忘れてはならないのは、設計/製品バリデーション≠プロセスバリデーションということです。規制当局は、設計/製品バリデーションとプロセスバリデーションの両方を個別に要求しているため、薬事申請の際には両方を同等に考慮する必要があります。
開発プロセスのどの段階でバリデーションを考えるべきか?医療機器メーカーは、バリデーション作業を始めるのに早すぎるということはないことを理解すべきです。企業は、正しい道を進んでいるか、正しい問題を解決しているかを把握するために、遅かれ早かれバリデーションを始めるべきです。
バリデーション(V&V)は反復的なプロセスであるため、計画が不十分だと良い投資ができません。テスト戦略をしっかりと定義することで、コストとテスト期間を最適化し、製品を期限内に市場投入できるようにすることができます。
テスト戦略の複雑さは、使用する技術や地理的なターゲット市場によって異なります。テスト戦略は、少なくとも以下の6つのパラメータをカバーする必要があります。
- 対象となる地域と関連する基準
- Time to market;
- バージョンアップして踏襲する規格です。
- テストラボ - 内部または独立したラボ。
- テストの順序を決める。
- テスト結果の提示
したがって、検証・妥当性確認のプロセスで使用されるテストも検証される必要があります。これは、テストが間違っていると、ユーザビリティや機能性のアウトプットが間違っているため、測定すべきものを確実に測定するためです。医療機器メーカーは、関連する規制に準拠した、効果的で文書化されたV&Vを必要としています。
医療機器の設計。リスクマネジメント
リスク移行戦略vs.リスク管理計画
国際的に認められたコンプライアンス基準に基づき、医療機器のリスク管理を実施しています。 ISO 14971:2007 医療機器-「医療機器へのリスクマネジメントの適用".また、リスクマネジメントの方針は、医療機器の設計・開発のすべての段階で取り入れられる必要があり、設計管理の側面にも関連していなければなりません。 [10].
リスクマネジメントに終わりはありません(少なくとも理論上は!)。リスクマネジメントの哲学は、堅苦しいルールにこだわるべきではないということです。リスクマネジメントとリスク移行戦略は、リスクマネジメントの意図を理解し、論理的かつ体系的にプロセスにアプローチすることです。言い換えれば ルールを守るだけでなく、考えてください。
Considering the complexity of medical device design, focused risk management practices help ensure usability, safety, and 規制遵守. It is a process of identifying, controlling, and preventing the failure that may cause hazards to users. It also mandates identifying associated risks.
図3は、リスクマネジメントのプロセスに関わるすべてのステップを示しています。このプロセスは、ハザードの特定から始まり、ハザードの結果とリスクの可能性に基づいて、関連するリスクが測定されます。
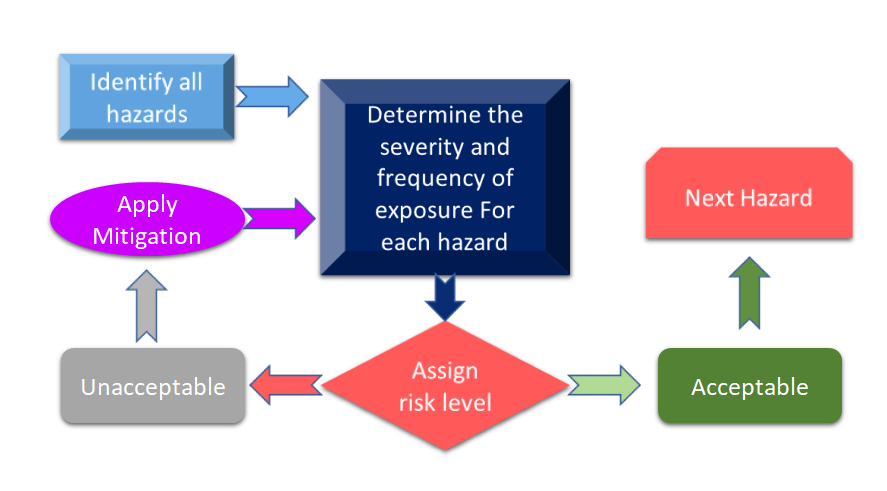
図3. 医療機器のリスクマネジメントプロセス (Adapted from) [11]).
医療機器のリスク管理プロセスで特定されたリスクが、定義された基準を超えている場合、リスク軽減が必要となります。リスクのレベルは、機器、技術、場合によっては貴社のリスク軽減プロセスの扱い方など、いくつかのパラメータに左右されますが、これらに限定されるものではありません。どのような規格が適用できるかを確認するために、機器のハザード分析を行うことが常に推奨されます。最近のISO 14971の改訂ではInternational Standard for Risk Management of Medical Devices(医療機器のリスク管理に関する国際規格)」の最近の改訂版では、リスク分析と予備的ハザード分析(PHA)が医療機器の主要な要件として挙げられています。 [12].簡単に言えば、PHAは、リスク評価と管理のための最初の枠組みを提供するものであり、PHAはリスク分析とリスク評価の両方をカバーしている。定義によれば、PHAは、機器の構造材料(MoC)、機器に使用される部品や原材料、人と機器または手動のインターフェイス、使用環境、動作原理、その他の関連要因から形成されるハザード、有害性、あらゆる危険な状況のリストから構成される。 [13].
結論
最後に、医療機器のスタートアップから既存の組織に至るまで、規則を読むことで得られるものは何もないが、哲学を理解することで得られるものは大きいということを覚えておいてください。
肝心なのは、リスク分析や計画を立てるときです。
- 設計・開発プロセスの初期段階からずっと活用すべきです。
- しばしば新しい情報を得て、設計や開発のプロセスにフィードバックすることができる(現在/将来のデバイスの場合)。
- どんなに計画を立てても、すべての危険性やリスクを排除することはできません。(しかし、多くのリスクを軽減することができます!(ここで説明されている設計管理の考え方に従うことで、自動的にリスクを軽減することができます)
すべての医療機器の市場投入までのルートは、使用パターン、素材、ユーザーエクスペリエンス、規制など、考慮すべきさまざまな要因により複雑です。
困ったときは medical device design? Browse experienced メドテック業界のエキスパート のKolabtreeに登録したり、無料でプロジェクトを投稿して提案を受けることができます。
リファレンス&リソース
- https://www.welldoc.com/health-plans/
- https://ec.europa.eu/docsroom/documents/10337/attachments/1/translations
- FDA, 2005, Total Product Lifecycle, FDA-CDRH Presentation by CDRH Director Dr David Feigal, http://www.fda.gov/cdrh/strategic/presentations/ tplc.html.
- Pietzsch, Jan & Shluzas, Lauren & Paté-Cornell, Marie-Elisabeth & Yock, Paul & Linehan, John.(2009).Stage-Gate Process for the Development of Medical Devices.Journal of Medical Devices.3(2).
- IEC 60601-1の第3版のための規制戦略 2020年9月9日に取得。
- https://www.meddeviceonline.com/doc/an-introduction-to-international-medical-device-standards-0001
- https://www.fda.gov/files/drugs/published/Design-Controls—Devices.pdf
- Feigal DW.付録D.規制の枠組みが医療機器の開発と革新に与える影響(Impact of the Regulatory Framework on Medical Device Development and Innovation).米国医学研究所(Institute of Medicine)委員会 公衆衛生 Effectiveness of the FDA 510(k) Clearance Process; Wizemann T, editor. Public Health Effectiveness of the FDA 510(k) Clearance Process: Balancing Patient Safety and Innovation: Workshop Report. Washington (DC): National Academies Press (US); 2010. Appendix D, Impact of the Regulatory Framework on Medical Device Development and Innovation. Available from: https://www.ncbi.nlm.nih.gov/books/NBK209794/.
- 1997年、FDA CDRH 1997年、医療機器メーカー向けデザインコントロールガイダンス
- https://starfishmedical.com/blog/iso-134852016-section-7/?doing_wp_cron=1599995964.4528369903564453125000
- Teixeira, M. B., and Bradley, R., 2003, Design Controls for the Medical Device Industry, Marcel Dekker, New York.
- ISO 14971:2019 - 医療機器 - 医療機器へのリスクマネジメントの適用
- ISO/TR 24971:2020 - 医療機器 - ISO 14971の適用に関するガイダンス
このシリーズの全記事
医療機器の開発・設計のための決定版ガイド
医療機器開発。成功のための3つのヒント
医療機器のデザイン。The Essential, Step-by-Step Guide
医療機器の商品化:スケッチから発売までの9つのステップ
医療機器の商業化の課題をどう克服するか
医療機器のローンチ製品を市場に投入するための重要なステップ
医療機器の市販後調査。包括的なガイド
Kolabtree helps businesses worldwide hire freelance scientists and industry experts on demand. Our freelancers have helped companies publish research papers, develop products, analyze data, and more. It only takes a minute to tell us what you need done and get quotes from experts for free.

Unlock Corporate Benefits
• Secure Payment Assistance
• Onboarding Support
• Dedicated Account Manager
Sign up with your professional email to avail special advances offered against purchase orders, seamless multi-channel payments, and extended support for agreements.


