



Shrinidh Joshi, esperto di dispositivi medici on Kolabtree, provides a comprehensive guide to dispositivo medico design, design controls, validation & verification, regulatory requirements and risk management.
Nel articolo precedente, we took a look at the overview of the dispositivo medico development process from the ideation to the discovery phase. In this article, we will focus more on medical device design, design controls, and compliance.
Progettazione di dispositivi medici: Regolamenti e conformità IEC e ISO
Ormai sapete che per entrare sul mercato il vostro dispositivo medico ha bisogno di qualificare determinati requisiti e standard normativi. Gli standard dei dispositivi medici come la Commissione Elettrotecnica Internazionale (IEC) o l'Organizzazione Internazionale per la Standardizzazione (ISO) permettono ai produttori di dispositivi medici, ai progettisti, ai laboratori e a tutti gli altri fornitori di servizi di sviluppo di dispositivi medici come CDMO di ispezionare, valutare e mantenere i loro dispositivi e attrezzature secondo determinati standard di qualità e usabilità.
La (IEC ha pubblicato il primo standard di dispositivi medici nel 1970, IEC 60601-1. IEC 60601-1, Apparecchiature elettromedicali - Parte 1: Questo è lo standard riconosciuto a livello internazionale che affronta i requisiti generali per le apparecchiature e i dispositivi elettromedicali che coprono gli standard per la sicurezza di base e le prestazioni essenziali [4].
Il documento IEC 60601-1 è stato rivisto periodicamente per rimanere in linea con gli ultimi sviluppi medici e le innovazioni tecnologiche nel campo dei dispositivi medici. La modifica più recente è stata realizzata nel 2012 (emendamento 1 alla IEC 60601-1). Questo standard rivisto contiene i requisiti per la considerazione del fattore umano, la valutazione delle prestazioni essenziali dei dispositivi medici, l'usabilità e i comandi. Include anche il software come dispositivo medico e specifica l'adozione di un ciclo di vita di sviluppo formale. Nell'ambito della IEC 60601-1 rivista sono incluse anche specifiche tecniche più recenti e riviste per i pericoli (sia elettrici che meccanici), i requisiti di etichettatura dei dispositivi medici (compresi i nuovi standard di etichettatura) e la documentazione.
Progettazione di dispositivi medici: Standard ISO
L'International Organization for Standardization ha anche delle specifiche per gli standard dei dispositivi medici. ISO 13485 e ISO 14971 sono standard ampiamente utilizzati in tutto il mondo per la gestione della qualità dei dispositivi medici. Oltre a questi standard internazionali, alcuni standard sono specifici della regione e tutti sono adottati dagli standard internazionali con poche modifiche e limitazioni.
Se un'azienda di dispositivi medici produce o vende dispositivi medici negli Stati Uniti, il dispositivo medico sarà regolato dalla FDA. L'American National Standards Institute (ANSI) è il rappresentante degli standard ISO negli Stati Uniti.
Ci sono altre due organizzazioni simili: l'Association for the Advancement of Medical Instrumentation (AAMI) e l'American Society for Quality (ASQ) che definisce gli standard per gli Stati Uniti.
Se un'azienda di dispositivi medici ha progettato un dispositivo considerando gli standard ISO, c'è anche la possibilità che la FDA non approvi il dispositivo. Poiché la FDA ha il proprio set di procedure per la gestione del rischio derivate da standard internazionali e regionali, che include:
(standard internazionale.)
- ANSI/AAMI/ISO 14971:2007 (R2010), Dispositivi medici - Applicazione della gestione dei rischi ai dispositivi medici (Uno standard regionale con aggiunte e modifiche rispetto allo standard internazionale di riferimento). [5].
Nel caso dello standard di gestione della qualità, non segue la versione internazionale o regionale dello standard ISO 13485. Questo perché la FDA ha linee guida diverse per la gestione della qualità nei dispositivi medici per il mercato statunitense.
Mentre, se l'azienda di dispositivi medici sta considerando l'Unione Europea, il Comitato Europeo di Standardizzazione (CEN) è la standardizzazione adottata da ISO e il Comitato Europeo di Standardizzazione Elettrotecnica (CENELEC) è lo standard regionale ispirato da IEC.
Il CEN è un po' modificato secondo i requisiti dell'ISO e scritto con un prefisso "EN". Per esempio:
- EN ISO 13485:2012, Dispositivi medici - Sistemi di gestione della qualità - Requisiti per scopi normativi.
- EN ISO 14971:2012, Dispositivi medici - Applicazione della gestione dei rischi ai dispositivi medici
I membri nazionali adottano questi standard dall'UE aggiungendo il loro prefisso. Per la Svizzera, Swiss Standards pubblica standard con "SN" come prefisso, come SN EN ISO 13485:2012 e SN EN ISO 14971:2012.
Nel caso del Canada, la Canadian Standards Authority (CSA) è l'organizzazione rappresentativa dell'ISO.
Regolamenti sui dispositivi medici e controllo della progettazione
I produttori di dispositivi medici devono seguire le linee guida del Design Control poiché gli enti normativi come FDA, Commissione Europea, Health Canada, e altri vogliono garantire che i dispositivi medici siano sicuri per i potenziali utenti prima che i produttori inizino a commercializzare i dispositivi. Come ho menzionato nella sezione precedente, la FDA non segue la ISO 13485 in quanto ha diversi requisiti per la gestione della qualità. I controlli di progettazione sono definiti sotto FDA 21 CFR 820.30 che ha un intento simile alla sezione 7.3 Progettazione e sviluppo descritta nelle linee guida per ISO 13485. Inoltre, la FDA incorpora i requisiti di Current Good Manufacturing Practice (cGMP) nella regolamentazione del sistema di qualità per seguire le buone pratiche di qualità per i progetti di dispositivi medici [6].
Il regolamento fornisce un quadro per implementare il controllo del design a un'ampia varietà di dispositivi. Il quadro offre flessibilità sia per le conformità normative che per il processo interno di progettazione e sviluppo.
Per implementare con successo il controllo del design dei dispositivi medici, sono necessari professionisti con un background sia tecnico che non tecnico, come l'amministrazione aziendale, la scienza della vita, l'ingegneria, l'informatica e le arti.
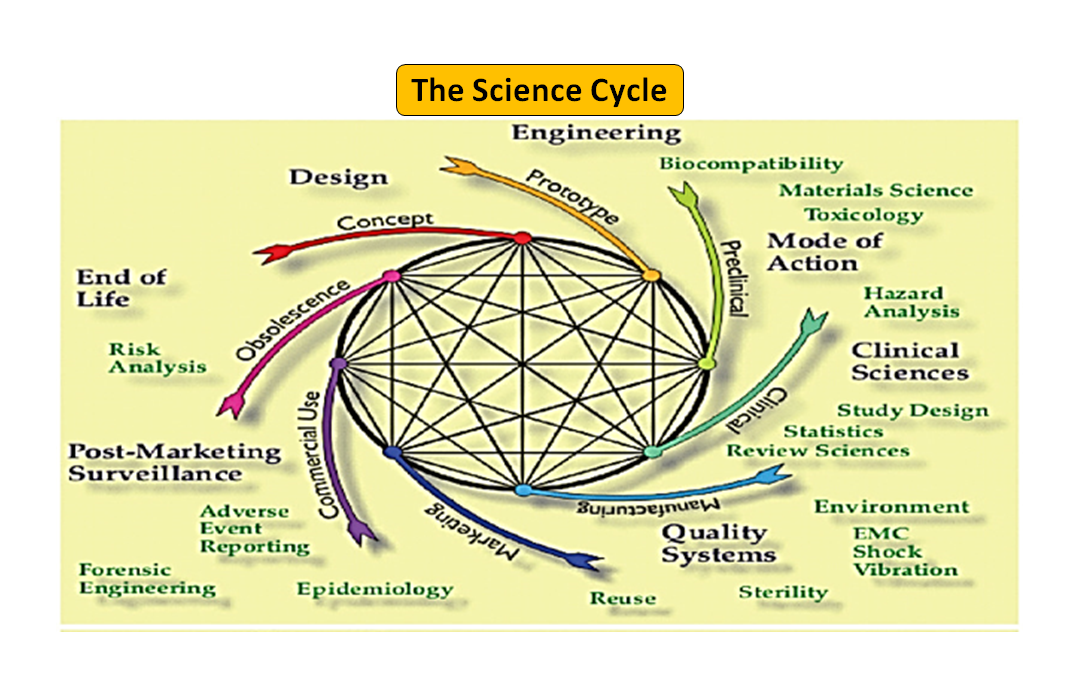
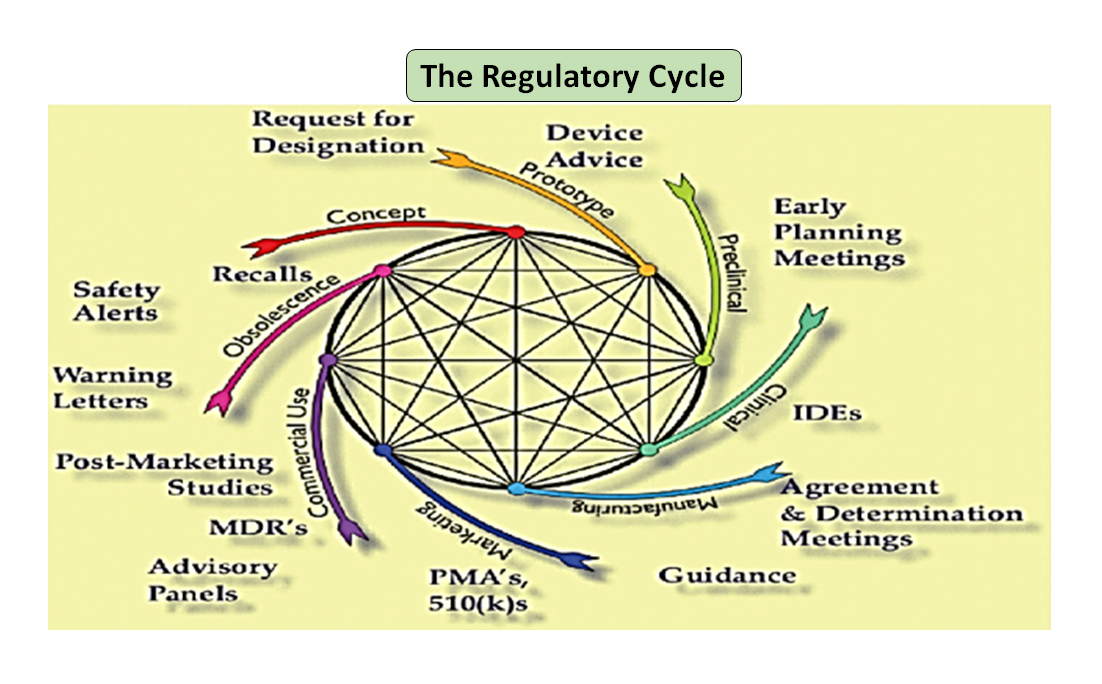
Figura 1: Ciclo di vita totale del prodotto. Il ciclo scientifico e il ciclo normativo (adattato da [7]).
È da notare che il ciclo di vita del dispositivo medico, dall'innovazione all'approvazione normativa e alla commercializzazione, è una serie di fasi interconnesse che guidano lo sviluppo del dispositivo (vedi Figura 1: Ciclo totale del prodotto). All'inizio, i prototipi progettati dai vostri ingegneri sono testati al banco per ottimizzare il design, testati per la biocompatibilità, gli estraibili, i dilavabili, la flessibilità o la resistenza generale del vostro dispositivo. Il ruolo del consulente normativo della vostra azienda è quello di sfogliare il database normativo per suggerirvi un documento guida che possa aiutarvi a determinare se il vostro prodotto sarà regolamentato come dispositivo medico o meno. L'uso previsto del vostro dispositivo medico e la sua modalità di funzionamento o di azione vi guideranno nella progettazione del dispositivo e decideranno anche il vostro percorso normativo se 510(k), PMA, De Novo, Pre-sub, IDE, HDE, master files ecc.
Come illustrato nella Figura 1, sia il processo scientifico che quello normativo si intrecciano lungo tutto il ciclo di vita del prodotto. Proprio come le diverse parti del ciclo di vita della scienza sono interconnesse, la scienza e i requisiti normativi sono intrecciati, ognuno informa e determina l'altro. C'è l'opportunità di costruire connessioni, sia alla FDA che nei produttori, in modo che le parti del ciclo di vita non rischino di essere considerate solo in modo isolato. Per esempio, non è raro che una domanda di pre-commercializzazione venga esaminata senza considerare l'esperienza post-commercializzazione di prodotti simili.
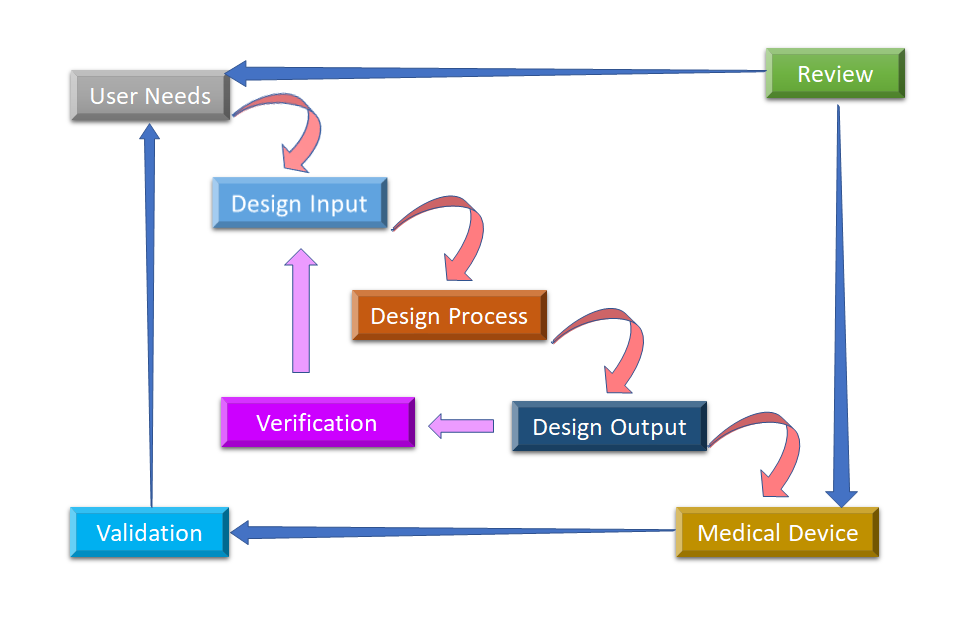
Figura 2: Processo di progettazione del flusso d'acqua per il controllo del design dei dispositivi medici (adattato da [8]).
La fase iniziale da cui parte il Design Control è lo sviluppo e l'approvazione del Design Input, che consiste nella progettazione del dispositivo e nei processi di fabbricazione da eseguire nella fase di produzione. Il controllo del design è un approccio olistico e non finisce con il trasferimento del design alla fase di produzione, una volta che il design è finalizzato. Ha anche un impatto sui processi di produzione in base ai cambiamenti nella fase di progettazione o anche al feedback post-produzione. È un processo continuo per sviluppare un prodotto che sia utilizzabile per un utente e quindi per il prodotto migliorato, considera i cambiamenti rivoluzionari dai modelli di utilizzo così come l'analisi dei prodotti falliti. Potete vedere nella Figura 2, come il Design Control può essere eseguito nel processo di design a cascata.
- Esigenze dell'utente:- I requisiti sono definiti considerando il bisogno del mercato e il dispositivo è progettato per rispondere a questo bisogno. Dopo una serie di evoluzioni, il design del dispositivo medico viene finalizzato e trasferito alla produzione per la fabbricazione. C'è bisogno di feedback durante ogni fase di questo processo.
- Input di progettazione: Questo è un processo iterativo. Quando un'organizzazione decide di affrontare una particolare necessità, rivede e testa l'accettabilità degli input di progettazione derivati dalla necessità. A quel punto, inizia il processo iterativo di conversione dei requisiti in design del dispositivo.
- Processo di progettazione: Questi input di progettazione sono convertiti in output di progettazione convertendo quei requisiti in specifiche di alto livello (che sono Design Output).
- Uscita del progetto: Il processo di verifica conferma se le specifiche soddisfano i requisiti o no. E l'output diventa l'input per rivedere i requisiti e questo processo va avanti fino a quando l'output del progetto è allineato con l'input del progetto.
- Dispositivo medico: Once the final design is ready, it is transmitted to the production facility for mass manufacturing. Design control regulation mandates Design History File (DHF), which illustrates the linkages and relationships between all the Design Controls and help to trace all changes throughout the entire sviluppo del prodotto processo.
Le aziende di dispositivi medici possono adottare un approccio basato su carta o su un software, sviluppato appositamente per il Design Control; i file della storia del design devono essere tracciabili e accessibili a tutti i membri del team.
Il diagramma di flusso qui sotto mostra un caso di studio per il controllo del design dei dispositivi medici.

Progettazione di dispositivi medici: Perché la tracciabilità è importante
Currently in the realm of the medical devices industry, it is an ideal practice to develop a traceability matrix that can illustrate the links and relations between user needs, design inputs and outputs, design verification and validation. When you are in the early phase for your device development you can maintain the device traceability using a spreadsheet or document version but as you move forward, its good idea to use cloud-based project management and document sharing platforms such as Microsoft Teams, Asana, Trello or whichever platform is suitable for your organization. The goal is as your project progresses you need to find an option which can save time because the old-school method of maintaining a traceability matrix might consume a lot of your time which you should rather be focusing on design verification and validation.
Una matrice di tracciabilità del Design Control è vitale per i team di sviluppo del prodotto, e specialmente per i project manager, perché la tracciabilità mostra la relazione e i collegamenti tra tutti i Design Control. Come si relazionano le esigenze dell'utente con gli input di progettazione? Come si collegano gli output di progettazione agli input di progettazione? Come si collegano le verifiche di progetto agli input di progetto e agli output di progetto? Come si collegano le convalide del progetto ai bisogni dell'utente? Una matrice di tracciabilità è uno strumento prezioso per mostrare una visione di alto livello e il flusso dello sviluppo del prodotto del dispositivo medico dall'inizio alla fine.
Gli sviluppatori di prodotti di best practice hanno fatto affidamento sulla tracciabilità dei Design Controls per molti, molti anni. E ora ISO 13485:2016 rende anche la tracciabilità un requisito. Come citato in ISO 13485:2016, 7.1 Pianificazione della realizzazione del prodotto, 1. c) le attività di verifica, convalida, monitoraggio, misurazione, ispezione e prova, manipolazione, stoccaggio, distribuzione e tracciabilità richieste specifiche per il prodotto insieme ai criteri di accettazione del prodotto; E 7.3.2 Pianificazione di progettazione e sviluppo, 1. e) i metodi per garantire la tracciabilità degli output di progettazione e sviluppo agli input di progettazione e sviluppo [9].
Progettazione di dispositivi medici: Verifica e convalida
Ogni dispositivo medico deve soddisfare gli obiettivi di funzionalità, usabilità e affidabilità per ottenere una quota di successo sul mercato.
Inoltre, i vostri stakeholder (pazienti, prescrittori, regolatori o utenti finali) presteranno attenzione alla sicurezza e all'efficacia del vostro dispositivo. È molto probabile che il vostro dispositivo sia progettato per rispondere a un bisogno insoddisfatto che può essere critico per la vita, per esempio un ventilatore o un dispositivo diagnostico che può rilevare le malattie cardiache. Pertanto, il test iterativo del vostro dispositivo con la verifica e la convalida è cruciale. Queste due fasi del processo di progettazione hanno lo scopo di confermare che il vostro dispositivo medico è in linea con i requisiti degli utenti e sta funzionando come previsto. In termini semplici, la verifica e la convalida del design possono garantire che il vostro dispositivo stia effettivamente facendo ciò che si suppone stia facendo. La verifica e la convalida del progetto servono anche a garantire i requisiti normativi, gli standard, la qualità del prodotto e il processo di produzione del vostro dispositivo medico. La verifica del progetto può valutare se il risultato del vostro progetto è conforme ai requisiti specificati, alle specifiche o ai requisiti normativi che sono specificati nell'input del progetto. D'altra parte, la convalida del progetto è intesa a valutare se il vostro dispositivo medico sta fornendo dei benefici basati sul bisogno degli utenti finali.
Design verifica chiede: "Abbiamo progettato bene il dispositivo?"
Design convalida chiede: "Abbiamo progettato il dispositivo giusto?"
I dispositivi medici possono consistere in diverse forme tecnologiche, dimensioni e diversi livelli di complessità. L'attività di verifica e convalida (V&V) è guidata dall'ambiente normativo e deve seguire gli standard internazionali. Le attività di V&V standardizzate possono snellire il processo di produzione e migliorare il processo di approvazione. Inoltre, i test automatizzati, le tecniche diagnostiche e gli strumenti di raccolta dati possono migliorare il processo di V&V [10].
- Convalida del prodotto vs. Convalida del processo
- Progettazione del dispositivo medico/convalida del prodotto:- Conformità alle esigenze dell'utente e del paziente, cioè il dispositivo funziona bene?
- Convalida del processo:- Il processo di produzione soddisfa le specifiche prestabilite.
È importante ricordare che la convalida del progetto/prodotto ≠ la convalida del processo. Le agenzie di regolamentazione richiedono sia la convalida del progetto/prodotto che la convalida del processo individualmente, quindi entrambi devono essere ugualmente presi in considerazione durante la presentazione normativa.
Quanto presto nel processo di sviluppo dovremmo pensare alla convalida? Un'azienda di dispositivi medici dovrebbe capire che non è mai troppo presto per iniziare il lavoro di convalida, un'azienda dovrebbe iniziare a convalidare prima che poi per capire che sta andando sulla strada giusta e risolvendo il problema giusto.
La convalida (anche V&V) essendo un processo iterativo consuma un buon investimento, se pianificato male. Una strategia di test ben definita può aiutare a ottimizzare i costi e il periodo di test per rendere il prodotto pronto per il mercato in tempo.
La complessità di qualsiasi strategia di test dipende dalle tecnologie da utilizzare e dai mercati geografici di destinazione. La strategia di test dovrebbe coprire almeno sei parametri menzionati di seguito:
- Geografie mirate e standard associati;
- Tempo di commercializzazione;
- Uno standard da seguire con una versione;
- Laboratori di prova - laboratori interni o indipendenti;
- Definire la sequenza dei test;
- Presentare il risultato del test
Di conseguenza, anche i test utilizzati per il processo di verifica e convalida devono essere convalidati. Questo per garantire che si misuri ciò che si deve misurare, perché un test sbagliato fornirà output errati di usabilità e funzionalità. Le aziende di dispositivi medici hanno bisogno di una V&V efficace e ben documentata, che sia conforme ai regolamenti associati.
Progettazione di dispositivi medici: Gestione del rischio
Strategia di migrazione del rischio vs. Piano di gestione del rischio
Le procedure di gestione del rischio per i dispositivi medici sono applicate secondo uno standard di conformità accettato a livello internazionale ISO 14971:2007 Dispositivi medici - "Applicazione della gestione del rischio ai dispositivi medici". Oltre a questo, le politiche di gestione del rischio devono essere incorporate in tutte le fasi della progettazione e dello sviluppo dei dispositivi medici e dovrebbero essere associate anche agli aspetti di controllo della progettazione [10].
La gestione del rischio non finisce mai (almeno in teoria!). La filosofia della gestione del rischio è che non dovrebbe riguardare un insieme duro e veloce di regole. La gestione del rischio e la strategia di migrazione del rischio riguardano la comprensione dell'intento della gestione del rischio e l'approccio al processo in modo logico e sistematico. In altre parole, non limitarti a seguire le regole... pensa!
Considering the complexity of medical device design, focused risk management practices help ensure usability, safety, and conformità normativa. It is a process of identifying, controlling, and preventing the failure that may cause hazards to users. It also mandates identifying associated risks.
La figura 3 mostra tutti i passi coinvolti nel processo di gestione del rischio. Il processo inizia con l'identificazione dei pericoli e poi il rischio associato viene misurato in base alle conseguenze dei pericoli e alla loro possibilità di rischio.
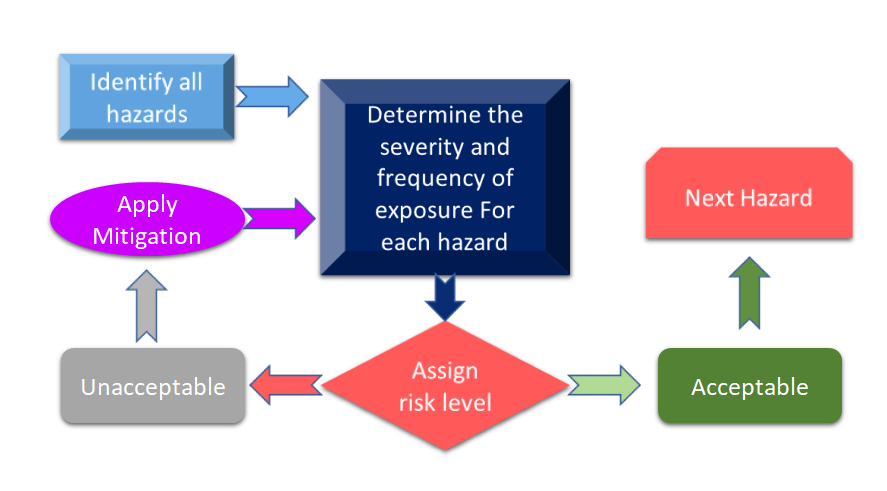
Figura 3: Processo di gestione del rischio per i dispositivi medici (adattato da [11]).
Nel caso in cui il rischio identificato durante il processo di gestione del rischio per i dispositivi medici sia superiore ai criteri definiti, allora sarà necessario avere una mitigazione del rischio. Il livello di rischio dipende da diversi parametri tra cui, ma non solo, il vostro dispositivo, le tecnologie e, in alcuni casi, il modo in cui la vostra azienda sta gestendo il processo di mitigazione del rischio. È sempre consigliabile condurre un'analisi dei rischi per il vostro dispositivo per vedere quali standard possono essere applicati al vostro dispositivo. Nella recente revisione di ISO 14971: International Standard for Risk Management of Medical Devices, l'analisi dei rischi e la Preliminary Hazard Analysis (PHA) sono identificati come requisiti primari per il vostro dispositivo medico [12]. In modo semplificato, PHA ha lo scopo di fornire il quadro iniziale per la valutazione e la gestione dei rischi e PHA copre sia l'analisi che la valutazione dei rischi. Come da definizione, PHA comprende un elenco di pericoli, danni, situazioni pericolose, formulate dai materiali di costruzione (MoC) dei vostri dispositivi, componenti o materie prime utilizzate nel vostro dispositivo, e interfacce uomo-dispositivo o manuali, ambiente di utilizzo, principio operativo e altri fattori rilevanti [13].
Conclusione
Alla fine, per ogni startup di dispositivi medici o per un'organizzazione consolidata, è importante ricordare che leggere i regolamenti non ti fa guadagnare nulla, ma capire la filosofia ti fa guadagnare molto!
In conclusione: quando si tratta di analisi e pianificazione dei rischi:
- Dovrebbe essere utilizzato all'inizio e durante tutto il processo di progettazione e sviluppo,
- Spesso ha generato nuove informazioni per il feedback nel processo di progettazione e sviluppo (à dispositivi attuali/futuri),
- Nessuna quantità di pianificazione può eliminare tutti i pericoli e i rischi... ma è possibile mitigarne molti! (Seguendo le filosofie di controllo della progettazione qui descritte, il rischio si riduce automaticamente!)
Il percorso di ogni dispositivo medico verso il mercato è complesso a causa dei vari fattori da prendere in considerazione come i modelli di utilizzo, il materiale, l'esperienza dell'utente, i regolamenti e altro ancora.
Ho bisogno di aiuto con medical device design? Browse experienced esperti dell'industria medtech su Kolabtree o pubblica il tuo progetto gratuitamente per ricevere proposte.
RIFERIMENTI E RISORSE
- https://www.welldoc.com/health-plans/
- https://ec.europa.eu/docsroom/documents/10337/attachments/1/translations
- FDA, 2005, Total Product Lifecycle, FDA-CDRH Presentation by CDRH Director Dr. David Feigal, http://www.fda.gov/cdrh/strategic/presentations/ tplc.html
- Pietzsch, Jan & Shluzas, Lauren & Paté-Cornell, Marie-Elisabeth & Yock, Paul & Linehan, John. (2009). Processo Stage-Gate per lo sviluppo di dispositivi medici. Giornale dei dispositivi medici. 3(2).
- Strategie normative per la terza edizione della IEC 60601-1 Recuperato il 9 settembre 2020.
- https://www.meddeviceonline.com/doc/an-introduction-to-international-medical-device-standards-0001
- https://www.fda.gov/files/drugs/published/Design-Controls—Devices.pdf
- Feigal DW. Appendice D. Impatto del quadro normativo sullo sviluppo e l'innovazione dei dispositivi medici. Comitato dell'Istituto di Medicina (USA) sul Salute pubblica Effectiveness of the FDA 510(k) Clearance Process; Wizemann T, editor. Public Health Effectiveness of the FDA 510(k) Clearance Process: Balancing Patient Safety and Innovation: Workshop Report. Washington (DC): National Academies Press (US); 2010. Appendix D, Impact of the Regulatory Framework on Medical Device Development and Innovation. Available from: https://www.ncbi.nlm.nih.gov/books/NBK209794/.
- 1997, FDA CDRH 1997, Design Control Guidance for Medical Device Manufacturers
- https://starfishmedical.com/blog/iso-134852016-section-7/?doing_wp_cron=1599995964.4528369903564453125000
- Teixeira, M. B., e Bradley, R., 2003, Design Controls for the Medical Device Industry, Marcel Dekker, New York.
- ISO 14971:2019 - Dispositivi medici - Applicazione della gestione dei rischi ai dispositivi medici
- ISO/TR 24971:2020 - Dispositivi medici - Guida all'applicazione della ISO 14971
Tutti gli articoli di questa serie:
Sviluppo e progettazione di dispositivi medici: Una guida definitiva
Sviluppo di dispositivi medici: 3 consigli per il successo
Progettazione di dispositivi medici: L'essenziale, guida passo dopo passo
Commercializzazione dei dispositivi medici: 9 passi dallo schizzo al lancio
Come superare le sfide della commercializzazione dei dispositivi medici
Lancio di un dispositivo medico: Passi chiave per portare il tuo prodotto sul mercato
Sorveglianza post-commercializzazione dei dispositivi medici: Una guida completa
Kolabtree helps businesses worldwide hire freelance scientists and industry experts on demand. Our freelancers have helped companies publish research papers, develop products, analyze data, and more. It only takes a minute to tell us what you need done and get quotes from experts for free.

Unlock Corporate Benefits
• Secure Payment Assistance
• Onboarding Support
• Dedicated Account Manager
Sign up with your professional email to avail special advances offered against purchase orders, seamless multi-channel payments, and extended support for agreements.


