



Il dottor Javier Quilez Oliete, un esperto freelance bioinformatics consultant on Kolabtree, provides a comprehensive guide to DNA sequencing analisi dei dati, including tools and software used to read data.
Introduzione
L'acido desossiribonucleico (DNA) è la molecola che porta la maggior parte delle informazioni genetiche di un organismo. (In alcuni tipi di virus, l'informazione genetica è trasportata dall'acido ribonucleico (RNA)). I nucleotidi (convenzionalmente rappresentati dalle lettere A, C, G o T) sono le unità di base delle molecole di DNA. Concettualmente, Sequenziamento del DNA è il processo di lettura dei nucleotidi che compongono una molecola di DNA (ad esempio, "GCAAACCAAT" è una stringa di DNA di 10 nucleotidi). Le attuali tecnologie di sequenziamento producono milioni di tali letture del DNA in un tempo ragionevole e ad un costo relativamente basso. Come riferimento, il costo del sequenziamento di un genoma umano - un genoma è l'insieme completo delle molecole di DNA in un organismo - è sceso il barriera $100 e può essere fatto in pochi giorni. Questo contrasta con la prima iniziativa di sequenziare il genoma umanoche è stato completato in un decennio e ha avuto un costo di circa $2,7 miliardi.
This capability to sequence DNA at high throughput and low cost has enabled the development of a growing number of sequencing-based methods and applications. For example, sequencing entire genomes or their protein-coding regions (two approaches known respectively as whole genome and exome sequencing) in disease and healthy individuals can hint to disease-causing DNA alterations. Also, the sequencing of the RNA that is transcribed from DNA—a technique known as RNA-sequencing—is used to quantify gene activity and how this changes in different conditions (e.g. untreated versus treatment). On the other side, chromosome conformation capture sequencing methods detect interactions between nearby DNA molecules and thus help to determine the spatial distribution of chromosomes within the cell.
Comune a queste e altre applicazioni del sequenziamento del DNA è la generazione di set di dati nell'ordine dei gigabyte e comprendenti milioni di sequenze lette. Pertanto, dare un senso agli esperimenti di sequenziamento ad alta produttività (HTS) richiede notevoli capacità di analisi dei dati. Fortunatamente, esistono strumenti computazionali e statistici dedicati e flussi di lavoro di analisi relativamente standard per la maggior parte dei tipi di dati HTS. Mentre alcune delle fasi (iniziali) di analisi sono comuni alla maggior parte dei tipi di dati di sequenziamento, l'analisi più a valle dipenderà dal tipo di dati e/o dall'obiettivo finale dell'analisi. Di seguito fornisco un primer sui passi fondamentali nell'analisi dei dati HTS e faccio riferimento a strumenti popolari.
Alcune delle sezioni seguenti sono focalizzate sull'analisi dei dati generati dalle tecnologie di sequenziamento a lettura breve (per lo più Illumina), poiché questi hanno storicamente dominato il mercato HTS. Tuttavia, le nuove tecnologie che generano letture più lunghe (ad es. Tecnologie Oxford Nanopore, PacBio) stanno guadagnando terreno rapidamente. Poiché il sequenziamento a lettura lunga ha alcune particolarità (ad esempio, tassi di errore più elevati), si stanno sviluppando strumenti specifici per l'analisi di questo tipo di dati.
Controllo di qualità (QC) delle letture grezze
L'analista impaziente inizierà l'analisi dai file FASTQ; il Formato FASTQ è stato a lungo lo standard per memorizzare i dati di sequenziamento a lettura breve. In sostanza, i file FASTQ contengono la sequenza nucleotidica e la per-base qualità di chiamata per milioni di letture. Anche se la dimensione del file dipende dal numero effettivo di letture, i file FASTQ sono tipicamente grandi (nell'ordine di megabyte e gigabyte) e compressi. Da notare che la maggior parte degli strumenti che usano i file FASTQ come input possono gestirli in formato compresso quindi, al fine di risparmiare spazio su disco, si raccomanda di non decomprimerli. Per convenzione, qui equiparerò un file FASTQ a un campione di sequenziamento.
FastQC è probabilmente lo strumento più popolare per effettuare il CQ delle letture grezze. Può essere eseguito attraverso un'interfaccia visiva o programmaticamente. Mentre la prima opzione può essere più conveniente per gli utenti che non si sentono a proprio agio con l'ambiente a riga di comando, la seconda offre una scalabilità e riproducibilità incomparabili (pensate a quanto noioso e soggetto a errori può essere eseguire manualmente lo strumento per decine di file). In entrambi i casi, l'output principale di FastQC è un file HTML reporting key summary statistiche about the overall quality of the raw sequencing reads from a given sample. Inspecting tens of FastQC reports one by one is tedious and it complicates the comparison across samples. Therefore, you may want to use MultiQCche aggrega i rapporti HTML da FastQC (così come da altri strumenti utilizzati a valle, ad esempio il trimming dell'adattatore, l'allineamento) in un unico rapporto.
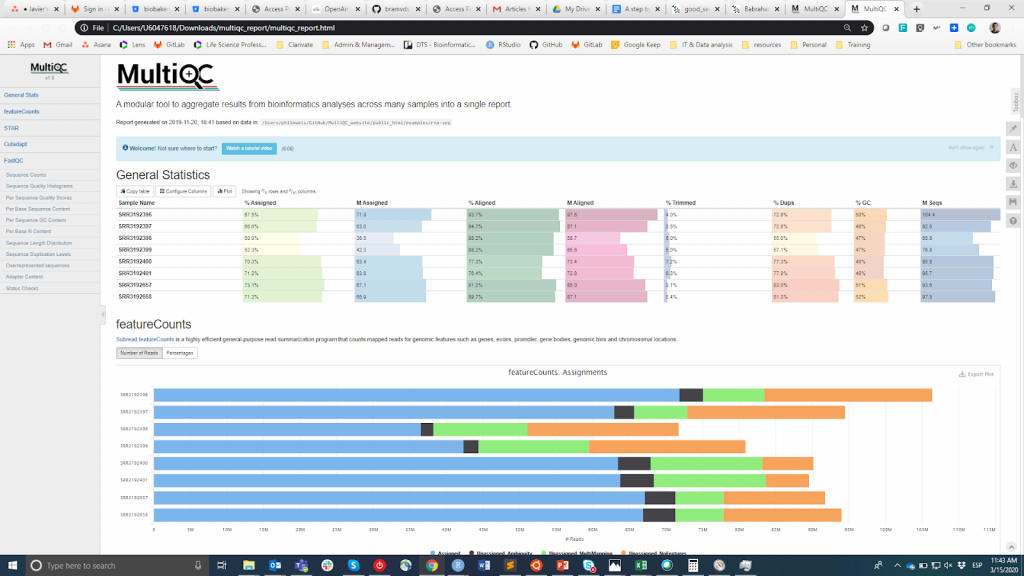
MultiQC
Le informazioni QC hanno lo scopo di permettere all'utente di giudicare se i campioni hanno una buona qualità e possono quindi essere utilizzati per le fasi successive o devono essere scartati. Sfortunatamente, non esiste una soglia di consenso basata sulla metrica FastQC per classificare i campioni come di buona o cattiva qualità. L'approccio che utilizzo è il seguente. Mi aspetto che tutti i campioni che sono passati attraverso la stessa procedura (ad esempio estrazione del DNA, preparazione della libreria) abbiano statistiche di qualità simili e una maggioranza di bandiere "pass". Se alcuni campioni hanno una qualità inferiore alla media, li userò comunque nell'analisi a valle tenendo conto di questo. D'altra parte, se tutti i campioni nell'esperimento ottengono sistematicamente "warning" o "fail" flag in più metriche (vedi questo esempio), sospetto che qualcosa sia andato storto nell'esperimento (ad es. cattiva qualità del DNA, preparazione della libreria, ecc.
Leggere il taglio
Il QC delle letture grezze aiuta a identificare i campioni problematici, ma non migliora la qualità effettiva delle letture. Per fare ciò, abbiamo bisogno di tagliare le letture per rimuovere le sequenze tecniche e le estremità di bassa qualità.
Le sequenze tecniche sono avanzi della procedura sperimentale (ad es. adattatori di sequenziamento). Se tali sequenze sono adiacenti alla vera sequenza della lettura, l'allineamento (vedi sotto) può mappare le letture nella posizione sbagliata nel genoma o diminuire la fiducia in un dato allineamento. Oltre alle sequenze tecniche, potremmo anche voler rimuovere sequenze di origine biologica se queste sono altamente presenti tra le letture. Per esempio, procedure di preparazione del DNA non ottimali possono lasciare un'alta percentuale di RNA ribosomiale (rRNA) convertito in DNA nel campione. A meno che questo tipo di acido nucleico è l'obiettivo dell'esperimento di sequenziamento, mantenendo letture derivate da rRNA aumenterà solo il carico computazionale dei passaggi a valle e può confondere i risultati. Da notare che se i livelli di sequenze tecniche, di rRNA o di altri contaminanti sono molto alti, che probabilmente saranno già stati evidenziati dal QC, si potrebbe scartare l'intero campione di sequenziamento.
Nel sequenziamento a lettura breve, la sequenza del DNA è determinata un nucleotide alla volta (tecnicamente, un nucleotide ogni ciclo di sequenziamento). In altre parole, il numero di cicli di sequenziamento determina la lunghezza della lettura. Un problema noto dei metodi di sequenziamento HTS è il decadimento della precisione con cui vengono determinati i nucleotidi con l'accumularsi dei cicli di sequenziamento. Questo si riflette in una diminuzione complessiva della qualità di chiamata per base soprattutto verso la fine della lettura. Come accade con le sequenze tecniche, cercare di allineare le letture che contengono estremità di bassa qualità può portare a un posizionamento errato o a una scarsa qualità di mappatura.
Per rimuovere le sequenze tecniche/contaminanti e le estremità di bassa qualità, leggi gli strumenti di trimming come Trimmomatic e Cutadapt esistono e sono ampiamente utilizzati. In sostanza, tali strumenti rimuovono le sequenze tecniche (disponibili internamente e/o fornite dall'utente) e tagliano le letture in base alla qualità, massimizzando la lunghezza della lettura. Le letture che rimangono troppo corte dopo il trimming vengono scartate (le letture eccessivamente corte, ad esempio <36 nucleotidi, complicano la fase di allineamento in quanto queste probabilmente mapperanno su più siti nel genoma). Potresti voler guardare la percentuale di letture che sopravvivono al trimming, poiché un alto tasso di letture scartate è probabilmente un segno di dati di cattiva qualità.
Infine, di solito eseguo nuovamente FastQC sulle letture tagliate per verificare che questo passo sia stato efficace e abbia sistematicamente migliorato le metriche di QC.
Allineamento
Con eccezioni (ad esempio assemblaggio de novo), l'allineamento (detto anche mappatura) è tipicamente il passo successivo per la maggior parte dei tipi di dati e applicazioni HTS. L'allineamento delle letture consiste nel determinare la posizione nel genoma da cui deriva la sequenza della lettura (tipicamente espressa come cromosoma:start-end). Quindi, in questa fase si richiede l'uso di una sequenza di riferimento su cui allineare/mappare le letture.
La scelta della sequenza di riferimento sarà determinata da molteplici fattori. Per esempio, la specie da cui deriva il DNA sequenziato. Mentre il numero di specie con una sequenza di riferimento di alta qualità disponibile sta aumentando, questo potrebbe non essere ancora il caso per alcuni organismi meno studiati. In questi casi, potresti voler allineare le letture a una specie evolutivamente vicina per la quale è disponibile un genoma di riferimento. Per esempio, dato che non esiste una sequenza di riferimento per il genoma del coyote, possiamo usare quella del cane strettamente correlato per l'allineamento delle letture. Allo stesso modo, potremmo ancora voler allineare le nostre letture a una specie strettamente correlata per la quale esiste una sequenza di riferimento di qualità superiore. Per esempio, mentre il genoma del gibbone è stato pubblicato, questo è rotto in migliaia di frammenti che non ricapitolano completamente l'organizzazione di quel genoma in decine di cromosomi; in questo caso, effettuare l'allineamento usando la sequenza umana di riferimento può essere utile.
Un altro fattore da considerare è la versione dell'assemblaggio della sequenza di riferimento, poiché vengono rilasciate nuove versioni man mano che la sequenza viene aggiornata e migliorata. È importante notare che le coordinate di un dato allineamento possono variare tra le versioni. Per esempio, più versioni del genoma umano possono essere trovate nel file UCSC Genome Browser. In ogni specie, favorisco fortemente la migrazione alla più recente versione dell'assemblaggio una volta che è completamente rilasciata. Questo può causare qualche fastidio durante la transizione, poiché i risultati già esistenti saranno relativi a versioni più vecchie, ma alla lunga ripaga.
Inoltre, anche il tipo di dati di sequenziamento è importante. Le letture generate da DNA-seq, ChIP-seq o protocolli Hi-C saranno allineate alla sequenza di riferimento del genoma. D'altra parte, poiché l'RNA trascritto dal DNA viene ulteriormente trasformato in mRNA (cioè gli introni rimossi), molte letture RNA-seq non riusciranno ad allinearsi a una sequenza di riferimento del genoma. Invece, abbiamo bisogno di allinearli alle sequenze di riferimento del trascrittoma o di usare allineatori consapevoli della divisione (vedi sotto) quando si usa la sequenza del genoma come riferimento. Correlata a questo è la scelta della fonte per l'annotazione della sequenza di riferimento, cioè il database con le coordinate dei geni, trascrizioni, centromeri, ecc. Di solito uso il database Annotazione GENCODE in quanto combina l'annotazione completa dei geni e le sequenze di trascrizione.
Una lunga lista di strumenti di allineamento di sequenze a lettura breve è stata sviluppata (vedi la sezione Allineamento di sequenze a lettura breve qui). Reviewing them is beyond the scope of this article (details about the algorithms behind these tools can be found qui). Nella mia esperienza, tra i più popolari ci sono Bowtie2, BWA, HISAT2, Minimap2, STAR e TopHat. La mia raccomandazione è che tu scelga il tuo allineatore in base a fattori chiave come il tipo di dati HTS e l'applicazione così come l'accettazione da parte della comunità, la qualità della documentazione e il numero di utenti. Ad esempio, si ha bisogno di allineatori come STAR o Bowtie2 che sono consapevoli delle giunzioni esone-esone quando si mappano RNA-seq al genoma.
Comune alla maggior parte dei mappatori è la necessità di indicizzare la sequenza usata come riferimento prima che l'allineamento effettivo abbia luogo. Questo passo può richiedere molto tempo, ma deve essere fatto solo una volta per ogni sequenza di riferimento. La maggior parte dei mappatori memorizza gli allineamenti in file SAM/BAM, che seguono il modello Formato SAM/BAM (I file BAM sono versioni binarie dei file SAM). L'allineamento è uno dei passi più impegnativi in termini di calcolo e di tempo nell'analisi dei dati di sequenziamento e i file SAM/BAM sono pesanti (nell'ordine dei gigabyte). Pertanto, è importante assicurarsi di avere le risorse necessarie (vedi la sezione finale sotto) per eseguire l'allineamento in un tempo ragionevole e memorizzare i risultati. Allo stesso modo, a causa delle dimensioni e del formato binario dei file BAM, evitate di aprirli con editor di testo; usate invece comandi Unix o strumenti dedicati come SAMtools.
Dagli allineamenti
Direi che non c'è un chiaro passo comune dopo l'allineamento, poiché a questo punto è dove ogni tipo di dati HTS e applicazione può differire.
Un'analisi a valle comune per i dati DNA-seq è la chiamata delle varianti, cioè l'identificazione delle posizioni nel genoma che variano rispetto al riferimento del genoma e tra gli individui. Un quadro di analisi popolare per questa applicazione è GATK per polimorfismo a singolo nucleotide (SNP) o piccole inserzioni/delezioni (indel) (Figura 2). Le varianti che comprendono pezzi più grandi di DNA (dette anche varianti strutturali) richiedono metodi di chiamata dedicati (vedi questo articolo per un confronto completo). Come per gli allineatori, consiglio di selezionare lo strumento giusto considerando fattori chiave come il tipo di varianti (SNP, indel o varianti strutturali), l'accettazione da parte della comunità, la qualità della documentazione e il numero di utenti.
Probabilmente l'applicazione più frequente di RNA-seq è la quantificazione dell'espressione genica. Storicamente, le letture dovevano essere allineate alla sequenza di riferimento e poi il numero di letture allineate a un dato gene o trascrizione veniva usato come proxy per quantificare i suoi livelli di espressione. Questo approccio di allineamento+quantificazione viene eseguito da strumenti come Gemelli, RSEM o featureCounts. Tuttavia, questo approccio è stato sempre più superato da nuovi metodi implementati in software come Kallisto e Salmone. Concettualmente, con tali strumenti la sequenza completa di una lettura non ha bisogno di essere allineata alla sequenza di riferimento. Invece, abbiamo solo bisogno di allineare abbastanza nucleotidi per essere sicuri che una lettura provenga da una data trascrizione. In parole povere, l'approccio di allineamento+quantificazione si riduce a un solo passo. Questo approccio è noto come pseudo-mapping e aumenta notevolmente la velocità della quantificazione dell'espressione genica. D'altra parte, tenete a mente che lo pseudo-mapping non sarà adatto per le applicazioni in cui è necessario l'allineamento completo (ad esempio la chiamata delle varianti dai dati RNA-seq).
Un altro esempio delle differenze nelle fasi di analisi a valle e gli strumenti necessari in tutta l'applicazione basata sul sequenziamento è ChIP-seq. Le letture generate con tale tecnica saranno utilizzate per il peak calling, che consiste nel rilevare le regioni nel genoma con un eccesso significativo di letture che indica dove la proteina target è legata. Esistono diversi peak caller e questa pubblicazione le indagini. Come ultimo esempio citerò i dati Hi-C, in cui gli allineamenti sono usati come input per strumenti che determinano le matrici di interazione e, da queste, le caratteristiche 3D del genoma. Commentare tutti i saggi basati sul sequenziamento va oltre lo scopo di questo articolo (per un elenco relativamente completo vedere questo articolo).
Prima di iniziare...
La parte restante di questo articolo tocca aspetti che possono non essere strettamente considerati come passi nell'analisi dei dati HTS e che sono ampiamente ignorati. Al contrario, io sostengo che è capitale che si pensi alle domande poste in Tabella 1 prima di iniziare ad analizzare i dati HTS (o qualsiasi tipo di dati), e ho scritto su questi argomenti qui e qui.
Tabella 1
| Pensateci | Azione proposta |
| Avete tutte le informazioni del vostro campione necessarie per l'analisi? | Raccogliere sistematicamente i metadati degli esperimenti |
| Sarà in grado di identificare inequivocabilmente il suo campione? | Stabilire un sistema per assegnare ad ogni campione un identificatore unico |
| Dove saranno i dati e i risultati? | Organizzazione strutturata e gerarchica dei dati |
| Sarà in grado di elaborare più campioni senza soluzione di continuità? | Scalabilità, parallelizzazione, configurazione automatica e modularità del codice |
| Lei o qualcun altro sarà in grado di riprodurre i risultati? | Documentate il vostro codice e le vostre procedure! |
Come menzionato sopra, i dati grezzi HTS e alcuni dei file generati durante la loro analisi sono dell'ordine dei gigabyte, quindi non è eccezionale che un progetto che include decine di campioni richieda terabyte di memoria. Inoltre, alcuni passi nell'analisi dei dati HTS sono computazionalmente intensivi (per esempio l'allineamento). Tuttavia, l'infrastruttura di stoccaggio e di calcolo necessaria per l'analisi dei dati HTS è una considerazione importante e spesso viene trascurata o non discussa. Come esempio, come parte di una recente analisi, abbiamo esaminato decine di articoli pubblicati che eseguono analisi di associazione a livello di fenoma (PheWAS). Le moderne PheWAS analizzano 100-1.000s sia di varianti genetiche che di fenotipi, il che comporta un'importante archiviazione di dati e potenza di calcolo. Eppure, praticamente nessuno dei documenti che abbiamo esaminato ha commentato l'infrastruttura necessaria per l'analisi PheWAS. Non sorprende che la mia raccomandazione sia quella di pianificare in anticipo i requisiti di archiviazione e di calcolo che si dovranno affrontare e condividerli con la comunità.
Hai bisogno di aiuto per analizzare i dati di sequenziamento del DNA? Mettiti in contatto con freelance bioinformatics specialist e esperti di genomica su Kolabtree.
Kolabtree helps businesses worldwide hire freelance scientists and industry experts on demand. Our freelancers have helped companies publish research papers, develop products, analyze data, and more. It only takes a minute to tell us what you need done and get quotes from experts for free.

Unlock Corporate Benefits
• Secure Payment Assistance
• Onboarding Support
• Dedicated Account Manager
Sign up with your professional email to avail special advances offered against purchase orders, seamless multi-channel payments, and extended support for agreements.


