



An exhaustive guide to cumplimiento de la normativa for IVD manufacturers, written by Sundeep Agarwal, con experiencia Consultor IVDR.
¿Qué es el IVDR?
The European Commission’s (EC) In Vitro Diagnostic Regulation (EU IVDR 2017/746) is a ‘legislative framework’ and a way forward towards global IVD safety, which assures that only reliable and effective IVDs are in the market. The European Commission is trying its best to make the salud system safer and error free in terms of diagnosis or outcomes.
Los productos sanitarios de diagnóstico in vitro (IVDD), 98/79/CE era una directiva mientras que IVDR es una legislación (reglamento) aplicable a todos los Operadores Económicos (OE), es decir, a los fabricantes, importadores, usuarios, organismos notificados y autoridades nacionales del Espacio Económico Europeo (EEE) y a los fabricantes y proveedores no pertenecientes a la UE que comercializan o tienen previsto distribuir DIV en el mercado europeo.
El IVDR consta de 113 artículos (10 capítulos) y quince anexos, en comparación con los 24 artículos y diez anexos del IVDD. No cabe duda de que el IVDR es un reglamento extenso y considerablemente estricto, pero la parte buena es que es más transparente en cuanto a los cambios y requisitos reglamentarios.
Hace hincapié en el enfoque basado en el ciclo de vida. Se aplicará a partir del 26 de mayo de 2022 y se espera que los operadores económicos (incluidos los fabricantes de fuera de la UE) se preparen de forma proactiva para la planificación y la aplicación de la misma. Todas las partes interesadas en el proceso serán ahora igualmente responsables del mercado de diagnósticos in vitro del Espacio Económico Europeo (EEE).
- Lo primero y más importante que debe hacer una organización es organizar un programa de formación (en línea o in situ, según sea el caso) sobre el Reglamento Europeo de Medicamentos para que todos los miembros de la organización sean conscientes de los cambios necesarios.
- Debe seguirse una comunicación oficial a todos los proveedores, subcontratistas o prestadores de servicios sobre el proceso y sus obligaciones.
- Realizar una evaluación de las deficiencias para comprobar la disponibilidad de sus recursos, un equipo competente para actualizar la documentación técnica requerida en virtud del IVDR de la UE. Tener la certificación ISO 13485: 2016 sería una ventaja añadida para establecer la conformidad.
- Es aconsejable (si es necesario) contratar a un experto en la materia o a un consultor externo desde la primera fase de la transición porque "una puntada a tiempo ahorra nueve".
- Este blog proporcionará un resumen detallado y consejos prácticos para cumplir con las expectativas de los organismos notificados y las autoridades competentes, tal como se describe en los diversos artículos y anexos bajo el IVDR 2017/746 de la UE.
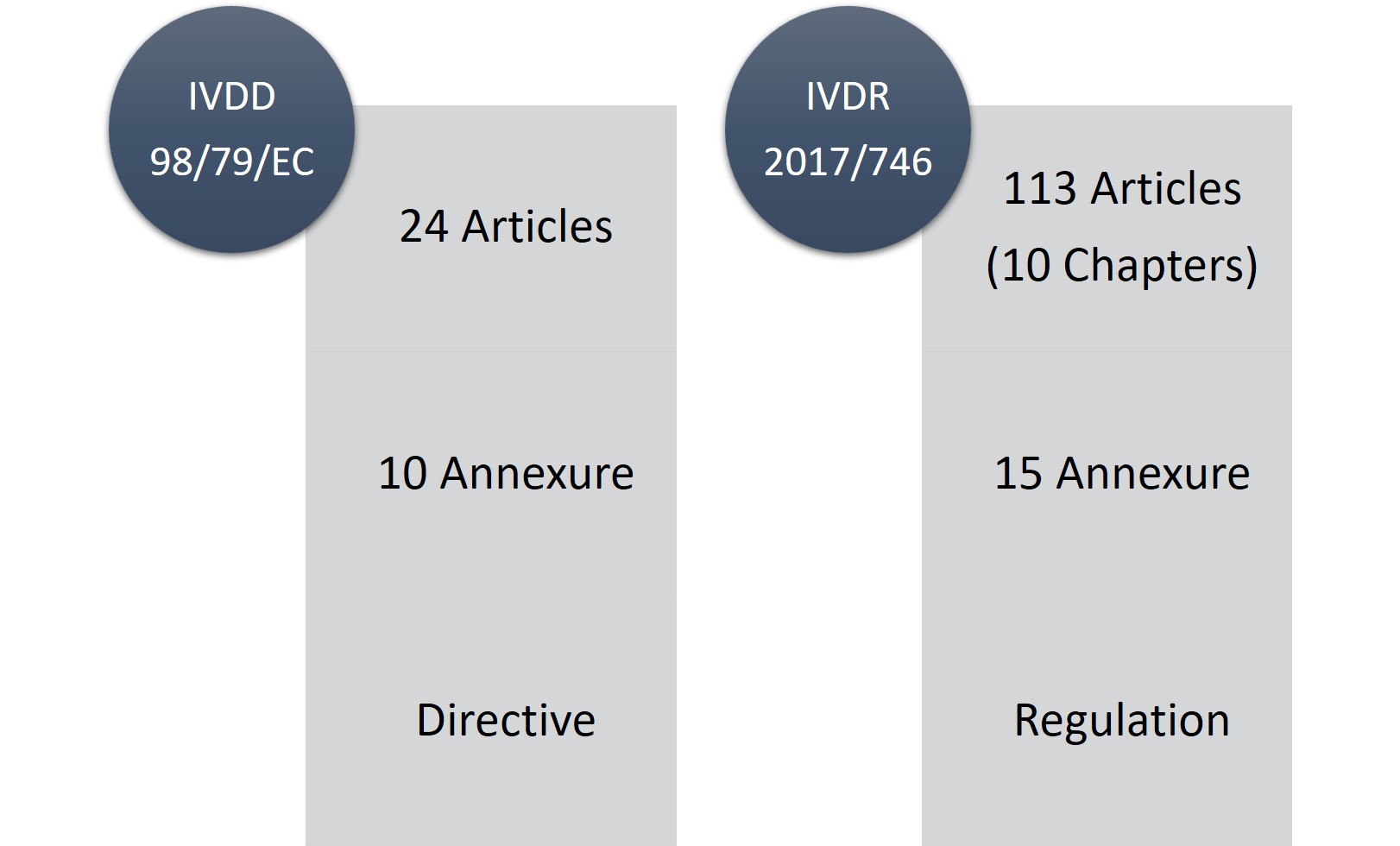
Figura1: IVDD frente a IVDR
1. Preparación para el cumplimiento del IVDR y los cambios comerciales
La decisión empresarial más importante para una organización sería concluir si quiere seguir colocando su DIV en el Espacio Económico Europeo (EEE). Si la respuesta es "sí", hay que obtener lo antes posible estimaciones (coste), plazos, alcance de la auditoría, código de producto, etc. de un organismo notificado. El paso de una directiva a un reglamento exige un cumplimiento obligatorio y una documentación técnica sólida para establecer la seguridad y la eficacia y conseguir la certificación CE. El IVDR se basa mucho más en las pruebas clínicas es decir, la validez científica, el rendimiento analítico y el rendimiento clínico para establecer la seguridad y la eficacia.
La participación de un organismo notificado (ON) en el proceso de certificación CE será una característica destacada del reglamento. Esto también indica una inversión adicional para el operador económico que puede aumentar indirectamente el coste del producto.
El nombramiento de un "Persona responsable del cumplimiento de la normativa (PRRC)" in accordance with Article 15 of EU IVDR 2017/746 is now mandatory; who shall assure the conformity of QMS, declaration of conformity, technical documentation, post market surveillance and reporting of adverse events are in compliance to EU IVDR.Manufacturers should ensure that the entire transition (including new certification application) is completed before the expiry of their existing IVDD Certificate or Self-certified Declaration of conformity. Certificates issued by notified bodies in accordance with IVDD 98/79/EC from 25 May 2017 shall become invalid after 27 May 2024. Be aware of the new timeline for application as per the EC official press release [1] dtd.20th Diciembre de 2022.
2. Comprensión clara de la clasificación
Reconsidere la nueva norma de clasificación según el anexo VIII del IVDR y compruebe si ha afectado a su clasificación anterior.
Realizar una clasificación correcta es esencial antes de preparar el proceso de certificación CE. A menos que seamos capaces de hacerlo, la ruta de conformidad será poco clara y retrasará o invalidará nuestros esfuerzos para cumplir con los requisitos del IVDR.
El IVDR es un enfoque basado en el riesgo para clasificar los productos con mayores controles de los organismos notificados y de las autoridades competentes. El Reglamento identifica cuatro clases de riesgo: Clase A (riesgo más bajo), Clase B, Clase C y Clase D (riesgo más alto), mientras que el Anexo VIII define siete normas de clasificación para clasificar correctamente los productos. Una característica única del IVDR es que los programas informáticos también se clasifican con arreglo a la regla de aplicación 1.4 del anexo VIII, que establece que "los programas informáticos que accionen un producto o influyan en su utilización deberán pertenecer a la misma clase que el producto. Si el software es independiente de cualquier otro producto, se clasificará por derecho propio[2]". Esto indica el alcance del software que debe ser regulado bajo el IVDR. Y el fabricante también tiene que realizar la verificación y validación del software (Anexo II, 6.4) en consecuencia.
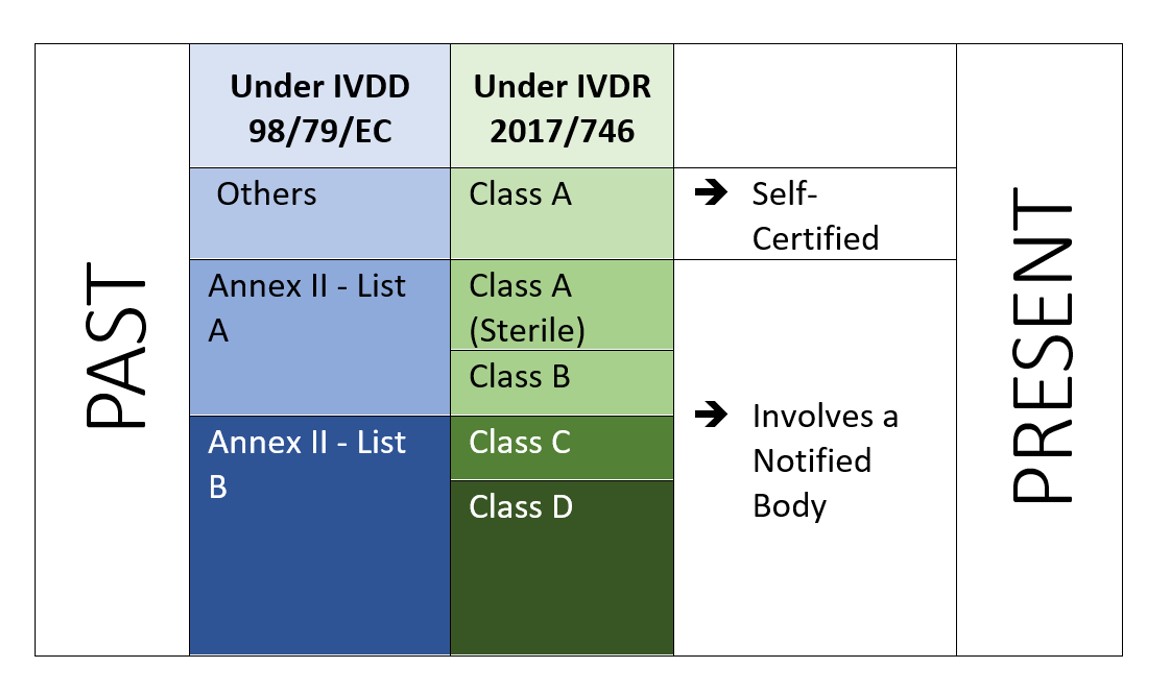
Figura 2: Clasificación basada en el riesgo según el IVDR 2107/746
3. Participación del organismo notificado
El papel de un organismo notificado (ON) sería uno de los elementos centrales y, por tanto, un mayor número de fabricantes tendría que ser auditado y certificado por un organismo notificado frente al método tradicional de "autocertificación".Los Operadores Económicos deben decidir cuidadosamente la ruta de evaluación de la conformidad (Anexos IX, X, XI del IVDR de la UE[3]).
El IVDR no sólo exige una inversión adicional, sino que tiene que asegurar que su documentación técnica y su sistema de gestión de la calidad cumplen los nuevos requisitos del IVDR. Bajo el IVDD, la mayoría de los DIVs son auto-certificados (92%), y no requieren la participación de un Organismo Notificado (excepto 8% del total de DIVs en el mercado[4]). Mientras que con el nuevo IVDR, el escenario no es el mismo.
Según un estudio "El impacto de las nuevas normas europeas de clasificación de DIV en la participación de los organismos notificados" por el Instituto Nacional de Salud pública y el Medio Ambiente, Bilthoven (Países Bajos) RIVM Letter report 2018-0082, A. van Drongelen et al., casi 85% de todos los DIVs requerirán la participación de un Organismo Notificado, dejando sólo 15% de DIVs elegibles para la autocertificación[5]..
Esto también significa que los fabricantes de productos de diagnóstico in vitro (IVD) experimentarán un cambio importante para cumplir con el nuevo proceso de clasificación y certificación. Además, dependiendo del uso previsto de los dispositivos y de la clase de riesgo, el fabricante deberá identificar un organismo notificado que pueda auditarlos y certificar sus productos. Los DIV de mayor riesgo (clase D) requerirán un laboratorio de referencia de la UE o paneles de expertos para verificar la declaración de rendimiento, además de la participación de un organismo notificado (ON) o una autoridad competente (AC). En la actualidad sólo hay seis organismos notificados designados en el marco del IVDR de la UE. No espere a iniciar su proceso de solicitud para evitar retrasos inesperados debido a la falta de disponibilidad de un organismo notificado.

Figura 3: Lista de organismos notificados designados en el marco del IVDR[6]
4. Establecimiento de un sistema de gestión de la calidad (SGC)
Se espera que los fabricantes de DIVs establezcan un sistema de gestión de la calidad (SGC) sólido y fiable en sus instalaciones. Es una obligación general de un fabricante según el artículo 10 del IVDR. El sistema de gestión de la calidad es un requisito esencial entre otros, sin el cual un fabricante no podrá obtener la aprobación.
QMS is to ensure that manufacturing, change control, customer complaints, resource management, supplier &sub-contractors’ controls and validation, performance evaluation, quality test, UDI Labelling, Post market surveillance etc. are according to approved QMS and Post Market Surveillance (PMS) plans.
El PRRC tiene que asegurarse de que el fabricante ha cumplido los requisitos del artículo 10 para "autocertificar" (emisión de la declaración de conformidad de acuerdo con el anexo IV) el DIV de clase A cuando no se requiere un organismo notificado (ON) en el proceso.
5. Prepárese para la interrupción de la cadena de suministro
Throughout the world, manufacturer depends largely on their supply chain and raw material to produce and deliver IVDs that are safe, accurate, and effective forthe intended use. Hence regulatory and quality concerns are also evolving to a higher level when it comes to the suppliers and sub-contractors’ controls. Manufacturer are therefore expected to proactively communicate the supply chain about their obligations and responsibilities of the suppliers and subcontractors. Legal manufacturer shall demonstrate adequate supplier control and monitoring, assure the supply chain is in compliance to the regulatory aspects of IVDR, reconsider the need for data integrity and quality of supplier data, implement robust supplier risk management and performance monitoring and periodically audit the supplier based on the associated risk to the finished products. Los reguladores y los organismos notificados están haciendo hincapié en que los fabricantes legales documenten claramente el nivel de los controles de los proveedores y demuestren con pruebas que tienen el potencial de mitigar el riesgo del producto o servicio suministrado por el proveedor.
6. Garantizar la preparación para las auditorías e inspecciones
De acuerdo con el artículo 88 del IVDR, Actividades de Vigilancia del Mercado, las autoridades competentes llevarán a cabo tanto inspecciones anunciadas (sin previo aviso) en los locales de los operadores económicos como de los proveedores y/o subcontratistas y, cuando sea necesario, en las instalaciones de los usuarios profesionales. Mientras que el fabricante incluirá información sobre la identificación de todos los sitios, incluidos los proveedores y subcontratistas, en los que se realicen actividades de fabricación en la documentación técnica de diseño y fabricación. Los organismos notificados (ON) que realicen la auditoría del SGC deberán identificar los vínculos entre los distintos centros de fabricación y sus proveedores y/o subcontratistas, así como la asignación de responsabilidades entre ellos. Esta información se tendrá en cuenta cuando el organismo notificado quiera auditar específicamente a alguno de esos proveedores o subcontratistas, o a ambos. Las instalaciones de los proveedores del fabricante, cuando se considere que afectan de forma significativa a la conformidad de los productos terminados, serán auditadas esencialmente por el organismo nacional (en particular cuando el fabricante no pueda demostrar un control suficiente sobre sus proveedores).
7. Plan para gestionar las auditorías sin previo aviso
En el marco de la supervisión posterior a la certificación, el organismo nacional procederá a realizar auditorías in situ sin previo aviso a los fabricantes y a sus subcontratistas o proveedores que realicen ensayos de productos y a supervisar el cumplimiento de cualquier condición que vincule a los fabricantes y que esté asociada a las decisiones de certificación, como la actualización de los datos clínicos a intervalos definidos.Además, tl organismo notificado realizará al azar, al menos una vez cada cinco años, auditorías sin previo aviso en las instalaciones del fabricante y, en su caso, en las de los proveedores y/o subcontratistas del fabricante, que podrán combinarse con la evaluación periódica de la vigilancia.
8. Reforzar las actividades de vigilancia posterior a la comercialización
Se recomienda encarecidamente a los fabricantes que refuercen sus requisitos de vigilancia posterior a la comercialización y que desarrollen un mecanismo de coordinación entre los Estados miembros de la UE en materia de vigilancia y control del mercado. En el marco de la evaluación de la vigilancia aplicable a los productos de la clase C y la clase D (anexo IX), el organismo notificado realizará periódicamente, al menos una vez cada 12 meses, las auditorías y evaluaciones pertinentes. Incluirá auditorías en los locales del fabricante y de los proveedores y/o subcontratistas, según proceda. El fabricante desarrollará esencialmente un procedimiento de registro y notificación de incidentes y de acciones correctivas de seguridad en el campo (FSCA).
9. Identificador único de dispositivo (UDI) y EUDAMED
El fabricante tendrá que establecer un sistema de UDI para identificar y facilitar la trazabilidad de los productos. El "Identificador del producto" y el "Identificador de la producción" deberán figurar en las etiquetas para mejorar la trazabilidad en el mercado de la UE. Se puede consultar una lista de entidades emisoras acreditadas, como GS1, HIBCC, ICCBBA, IFA GmbH, para gestionar un sistema de asignación de UDI. En la actualidad, las EI mencionadas son válidas desde el 27th junio de 2019, pero será conveniente confirmar su validez mientras se toma una decisión definitiva sobre su aplicación.
La Base de Datos Europea de Productos Sanitarios (EUDAMED) proporcionará una visión general de todos los productos sanitarios disponibles en la Unión Europea. Consta de seis módulos relacionados con:
- Registro de actores,
- Identificación única de dispositivos (UDI) y registro de dispositivos,
- Organismos notificados y certificados,
- Investigaciones clínicas y estudios de rendimiento,
- Vigilancia y seguimiento postcomercialización, y
- Vigilancia del mercado.
Para garantizar una mayor transparencia a través de un EUDAMED completo, parte de la información de los operadores económicos será de acceso público. Mientras que la información confidencial sólo será accesible para el Operador Económico, los Patrocinadores, las Autoridades Notificadas y las Autoridades Competentes de los Estados miembros de la UE.
10. Requisitos para los "dispositivos internos"
Health institution developing ‘in-house devices’ (or ‘laboratory-developed tests’) which are meant to be used by the same health institution shall not be marketed or sold to other legal entity. Such devices may be used for the diagnosis and treatment, especially for rare diseases. The institution is expected to comply with only the requirement of Annex I of IVDR (general safety and performance requirements), and exempted from rest of the regulation until 26 May 2024; provided the health institution meets a number of conditions set out in Article 5(5) of the Regulation and has an appropriate quality management system, which complies to the international standard setting out the quality and competence requirements for medical laboratories (EN ISO 15189) or other national provisions, and is able to justify that target patient group’s specific needs cannot appropriately be met by an equivalent device available on the market.
Referencias
[1] Comunicado de prensa oficial de la CE del 20 de septiembreth Diciembre de 2021, implantación progresiva del Reglamento sobre productos sanitarios de diagnóstico in vitro. Se puede acceder a él en https://ec.europa.eu/commission/presscorner/detail/en/IP_21_6965 [2]REGLAMENTO (UE) 2017/746 DEL PARLAMENTO EUROPEO Y DEL CONSEJO de 5 de abril de 2017 sobre productos sanitarios para diagnóstico in vitro y por el que se deroga la Directiva 98/79/CE y la Decisión 2010/227/UE de la Comisión; ANEXO VIII NORMAS DE CLASIFICACIÓN, 1. NORMAS DE APLICACIÓN Punto 1.4 Página 304 [3]Reglamento (UE) 2017/746 del Parlamento Europeo y del Consejo, de 5 de abril de 2017, sobre productos sanitarios para diagnóstico in vitro y por el que se deroga la Directiva 98/79/CE y la Decisión 2010/227/UE de la Comisión.ANEXO IX Evaluación de la conformidad basada en un sistema de gestión de la calidad y en la evaluación de la documentación técnica, página 306, ANEXO X Evaluación de la conformidad basada en el examen de tipo, página 314, ANEXO XI Evaluación de la conformidad basada en el aseguramiento de la calidad de la producción, página 317
[4] Comunicado de prensa dtd. 14 de octubre de 2021, Bruselas; Salud pública: La Comisión propone un despliegue progresivo del nuevo Reglamento sobre productos sanitarios para diagnóstico in vitro [5] El impacto de las nuevas normas europeas de clasificación de DIVs en la participación de los organismos notificados: un estudio sobre los DIVs registrados en los Países Bajos; van Drongelen A, de Bruijn A, Pennings J, van der Maaden T 32 p en inglés 2018, informe de carta del RIVM 2018-0082 [6] La lista anterior se basa en los datos consultados el 5 de marzo de 2021. 5 de marzo de 2021, para las últimas actualizaciones de la lista, puede acceder al sitio web oficial de la CE en https://ec.europa.eu/growth/tools-databases/nando/index.cfm?fuseaction=directive.notifiedbody&dir_id=35Kolabtree helps businesses worldwide hire freelance scientists and industry experts on demand. Our freelancers have helped companies publish research papers, develop products, analyze data, and more. It only takes a minute to tell us what you need done and get quotes from experts for free.

Unlock Corporate Benefits
• Secure Payment Assistance
• Onboarding Support
• Dedicated Account Manager
Sign up with your professional email to avail special advances offered against purchase orders, seamless multi-channel payments, and extended support for agreements.


