



Presentaciones a la FDA consultor y redactor de normativas Samradni Patil proporciona una Lista de comprobación para la presentación de 510k to help dispositivo médico companies with quick and easy FDA clearance.
El proceso de presentación 510(k) se utiliza normalmente para que los dispositivos médicos de clase II obtengan la autorización de la Administración de Alimentos y Medicamentos de Estados Unidos (FDA). El proceso de aprobación previa a la comercialización (PMA) suele utilizarse para los productos sanitarios de clase III.
El proceso de revisión 510(k) determina la equivalencia sustancial (SE) con un dispositivo similar comercializado legalmente, también llamado dispositivo predicado. El producto debe ser al menos tan seguro y eficaz como el producto comercializado legalmente para poder afirmar que es sustancialmente equivalente a él. El producto sometido a la revisión 510(k) debe demostrar lo siguiente para poder afirmar que es equivalente al producto de referencia:
- Mismo uso previsto que el dispositivo comercializado legalmente (dispositivo predicado)predicado
- Las mismas características tecnológicas que el dispositivo predicado o
- No se plantean características tecnológicas e información/pruebas diferentes que sugieran que el dispositivo es tan seguro y eficaz como el dispositivo predecesor ni cuestiones diferentes sobre la seguridad y la eficacia que el dispositivo predecesor.
El incumplimiento de los criterios anteriores conduce a la determinación de Equivalencia No Sustancial (NSE).
Lista de comprobación para la presentación de 510k
El proceso de revisión de la FDA 510(k) puede dividirse, a grandes rasgos, en dos pasos.
- Revisión de la aceptación
- Examen de fondo
Revisar los plazos
| Tipo de revisión | Calendario (días naturales) | Resultado del proceso |
| Revisión de la aceptación | Para el día 15 | La FDA informa al solicitante si la solicitud es aceptada para Examen de fondo o Puesta en espera RTA |
| Examen de fondo | Para el día 60 | Revisión interactiva o Solicitud de información adicional |
Nota: El día 1 es el día en que la FDA recibe la solicitud 510(k)
En primer lugar, analicemos qué tipo de problemas afrontan las empresas de productos sanitarios durante estos procesos de revisión.
Revisión de la aceptación
Si el 510(k) no se acepta en esta fase, se coloca en Retención por rechazo (RTA). Según Datos de la FDA, en 2018 aproximadamente 30% 510(k)s fueron colocados en una retención RTA.
Examen de fondo
El siguiente gráfico muestra el porcentaje de solicitudes de información adicional emitidas por la FDA durante la fase de revisión sustantiva.
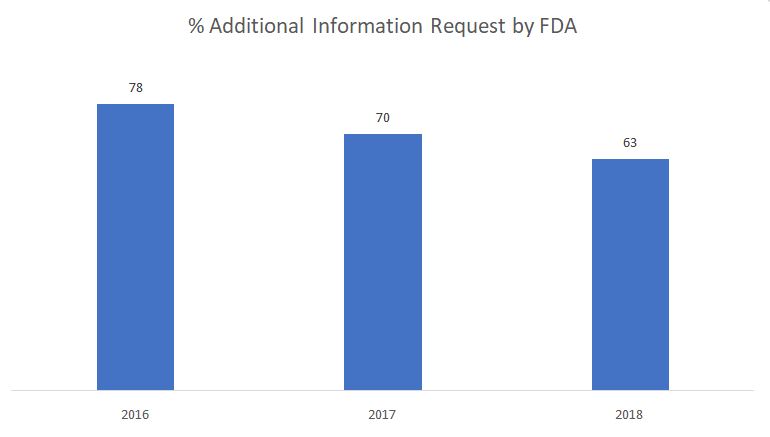
Fuente: FDA
El porcentaje de 510ks determinado como No sustancialmente equivalente (NSE) se muestra a continuación.
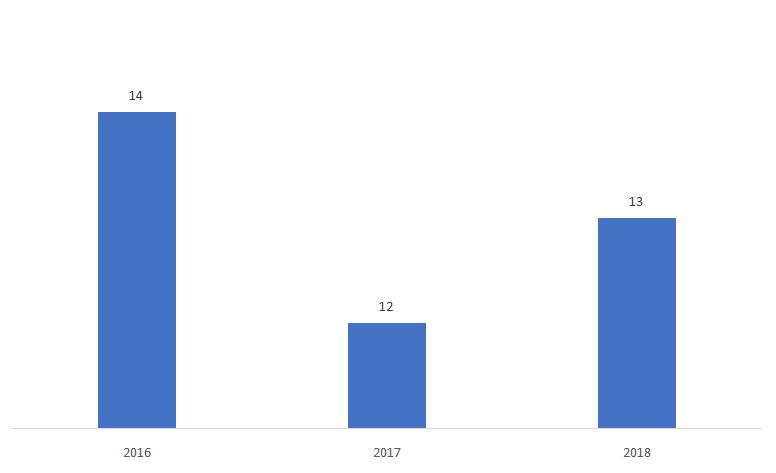
Fuente: FDA
Solicitudes de información adicional comunes de la lista de comprobación de la presentación de 510k
Ahora que entendemos las cuestiones comunes que se plantean como parte del proceso de revisión 510k, vamos a centrarnos en qué tipo de solicitudes de información adicional son comunes.
El Datos de la FDA muestra los siguientes tipos de solicitudes de información adicional:
- Descripción inadecuada del dispositivo
- Discrepancias a lo largo de la presentación - Las discrepancias en esta categoría suelen estar relacionadas con la descripción del dispositivo o las indicaciones de uso
- Problemas con las indicaciones de uso
- No seguir o no abordar los documentos de orientación actuales o las normas reconocidas
- Las pruebas de rendimiento requeridas para ciertos tipos de dispositivos faltan por completo (es decir, no se proporcionan datos de rendimiento en absoluto)
- Los datos clínicos requeridos para ciertos tipos de dispositivos faltan por completo (es decir, no se proporcionan datos clínicos en absoluto)
Ahora, hablemos de las mejores prácticas a seguir durante la preparación y presentación de una solicitud 510k.
Lista de comprobación para la presentación de 510k:
1. Carta de rechazo a la aceptación (RTA)
El propósito de la revisión de aceptación en la etapa inicial es comprobar si la solicitud 510k está administrativamente completa. Se recomienda encarecidamente revisar la lista de comprobación de la aceptación que figura en el documento de orientación "Política de rechazo a la aceptación de 510ks".
Para garantizar el éxito del examen de aceptación, se sugiere que cada empresa siga los elementos de la siguiente tabla del documento de orientación.
- Tabla de preguntas preliminares: Aunque esta lista de comprobación está pensada para que el revisor principal tome una determinación inicial, se recomienda encarecidamente responder a estas preguntas de manera informal antes de presentar la solicitud a la FDA.
- Tabla de elementos organizativos: Estos elementos ayudan a organizar la solicitud 510(k) de manera que se pueda identificar fácilmente la información en la solicitud 510(k).
- Elementos de una presentación completa (artículos RTA) Tabla: Las empresas deben prestar más atención a los elementos enumerados en este cuadro, ya que son fundamentales para no recibir una carta RTA.
2. Descripción inadecuada del dispositivo
La descripción del dispositivo es obligatoria en la solicitud 510(k). Se sugiere añadir una breve descripción y especificaciones técnicas en esta sección. Deben incluirse todos los modelos y accesorios del producto sanitario. Deben incluirse imágenes, diagramas, dimensiones, dibujos y tolerancias de cada componente. La omisión de un modelo o accesorio importante puede dar lugar a confusión y a preguntas adicionales. Unas especificaciones técnicas inadecuadas pueden dar lugar a un malentendido y a la solicitud de pruebas adicionales.
3. Información incoherente en toda la presentación
- Inconsistencia en la descripción del dispositivo: Si una empresa decide presentar el 510(k) para añadir un modelo adicional, es importante que las secciones aplicables, como la carta de presentación, la descripción del dispositivo, el etiquetado, la discusión sobre la equivalencia sustancial y las secciones relacionadas con el rendimiento, estén alineadas con el cambio real. La incoherencia puede dar lugar a retrasos administrativos y, en el peor de los casos, a la solicitud de pruebas adicionales.
- Inconsistencia en la indicación de uso: Al igual que la descripción del dispositivo, la incoherencia en la indicación de uso en varias secciones del 510(k) puede causar problemas. La declaración de la indicación de uso es muy importante para determinar la SE.
Las incoherencias en varias secciones podrían evitarse fácilmente mediante una revisión cuidadosa de la presentación antes de enviarla a la FDA. Siempre es una buena idea tener un par de ojos adicionales para echar un vistazo a varias secciones para evitar tales errores.
4. Uso previsto diferente al del dispositivo predicativo
Para obtener la determinación de SE por parte de la FDA, el dispositivo debe tener la el mismo uso previsto para el dispositivo predicado. Esto es importante porque un uso previsto diferente al del dispositivo predecesor puede dar lugar a problemas de seguridad y eficacia diferentes. En tal caso, el 510(k) puede no ser una vía adecuada para obtener la autorización del producto. Hay que tener en cuenta que las diferencias en la indicación de uso entre el producto predecesor y el producto sometido a la revisión del 510(k) no tienen por qué dar lugar a un uso previsto diferente. Las empresas deben hacer esfuerzos adicionales para demostrar claramente que las diferencias en la indicación de uso no han dado lugar a un uso previsto diferente.
La guía de la FDA "El programa 510(k): Evaluación de la equivalencia sustancial en las notificaciones previas a la comercialización [510(k)]." para aclarar las cuestiones relacionadas con el uso previsto.
5. Información inadecuada/falta de pruebas
- Información inadecuada sobre las pruebas:
Es importante comprender las pruebas aplicables a cualquier dispositivo en particular. Según el tipo de dispositivo, pueden ser necesarias pruebas de seguridad eléctrica, de compatibilidad electromagnética (CEM), de biocompatibilidad, de validación de software, de esterilización y de usabilidad para reclamar la seguridad y la eficacia de un dispositivo.
A menudo, las empresas infravaloran la cantidad de pruebas necesarias o intentan justificar que no se realicen determinadas pruebas
Ejemplo: Las empresas pueden basarse en los datos de biocompatibilidad de un dispositivo similar para reclamar la biocompatibilidad de su producto. Este enfoque puede ser aceptable en algunos casos. Sin embargo, muchas veces el proceso de fabricación entre estos dispositivos puede justificar pruebas de biocompatibilidad separadas en el dispositivo bajo la revisión 510(k).
Estas pruebas adicionales pueden llevar varias semanas. Si la FDA solicita la realización de estas pruebas adicionales durante la revisión del 510(k), se añade mucho tiempo en la autorización final del 510(k).
Appropriate teams should give thorough consideration to current FDA guidance, product design, risk management process to make determination about amount of testing required.
- Falta información sobre las pruebas:
Es importante entender la diferencia entre el 510(k) tradicional y el 510(k) especial. Todos los datos de las pruebas deben incluirse en la presentación tradicional del 510(k).
6. No seguir o abordar de otro modo Actual Documento(s) de orientación o normas reconocidas
Como ya se ha dicho, hay que asegurarse de que las pruebas se realizan para demostrar la conformidad con las últimas normas reconocidas. Puede haber cambios significativos en la última versión de la norma en comparación con la versión anterior. Esto puede dar lugar a preguntas adicionales sobre la seguridad y la eficacia si el dispositivo no se prueba con la última versión de la norma.
La consulta de los últimos documentos de orientación ayuda a comprender las expectativas o recomendaciones de la FDA. Esto también facilita el proceso de revisión de la FDA, ya que la presentación está escrita en un formato fácilmente comprensible para el revisor.
7. Determinación del NSE
El objetivo final del proceso 510(k) es determinar la equivalencia sustancial (SE) con un dispositivo anterior. Cuando la FDA solicita información adicional durante la fase de revisión sustantiva, las empresas deben revisar cuidadosamente cada solicitud y proporcionar una respuesta científicamente sólida. No proporcionar los datos/respuesta solicitados puede dar lugar a la determinación de NSE. Muchas determinaciones de NSE se deben a la falta de suministro de datos de rendimiento.
Es muy recomendable consultar y colaborar con la FDA para entender las expectativas en esta fase. He aquí otros errores comunes que he observado en mi experiencia.
Aspectos administrativos
8. Presentación de la solicitud en la dirección correcta
Se trata de un error humano fácilmente evitable. Consulte siempre el sitio web de la FDA para conocer la dirección correcta para presentar su solicitud.
9. Incluyendo una copia impresa y una copia electrónica según la última recomendación de la FDA
La FDA tiene requisitos específicos para el número de copias impresas y electrónicas que deben presentarse para la solicitud 510(k). Actualmente, se requiere 1 copia impresa y 1 copia electrónica. No haga ninguna suposición. Consulte el sitio web de la FDA antes de sacar cualquier conclusión.
10. Problemas relacionados con eCopy
El envío debe cumplir las normas técnicas de eCopy. Consulte el Guía de eCopy para preparar una copia electrónica de la solicitud.
Para evitar la carta de retención de eCopy es necesario seguir la convención de nombres de PDF y la recomendación de tamaño de los archivos. Aunque el uso de la Herramienta eSubmitter-eCopies es voluntario, esta herramienta ayuda a validar el eCopy según las recomendaciones de la FDA.
11. Envío de información al revisor
Después de recibir la carta de retención inicial de la FDA (retención RTA, retención eCopy), las empresas suelen cometer un error al enviar la información al revisor. Compruebe el correo electrónico recibido de la FDA o la guía apropiada de la FDA sobre dónde enviar la respuesta a la carta de retención.
Aspectos técnicos
12. 510(k) tradicional frente a 510(k) especial
Las empresas pueden cometer el error de clasificar la solicitud como 510(k) tradicional o 510(k) especial. La principal diferencia entre el 510(k) tradicional y el 510(k) especial es el tiempo necesario para que la FDA revise la solicitud. La 510(k) especial tarda 30 días naturales, mientras que la 510(k) tradicional tarda 90 días naturales. He visto varias veces que la FDA pide a las empresas que conviertan el 510(k) especial en 510(k) tradicional. En este caso, las empresas acaban perdiendo mucho tiempo en el proceso de conversión.
Según la FDA, un 510(k) especial puede ser apropiado cuando:
- La propuesta de modificación es presentada por el fabricante legalmente autorizado a comercializar el producto existente;
- Los datos de rendimiento son innecesarios, o si los datos de rendimiento son necesarios, se dispone de métodos bien establecidos para evaluar el cambio; y
- Todos los datos de rendimiento necesarios para respaldar la equivalencia sustancial pueden revisarse en un formato de resumen o de análisis de riesgos.
To avoid such mistake, do thorough investigación on the FDA database to identify if similar change was submitted as Special 510(k) or Traditional 510(k). Refer to the documentos de orientación de la FDA. Si todavía tiene dudas, pida ayuda a los consultores de reglamentación. Utilice un enfoque basado en el riesgo. Si todavía tiene dudas, se recomienda adoptar un enfoque conservador y presentar un 510(k) tradicional.
Desafíos únicos
13. Naturaleza del dispositivo
Algunos dispositivos pueden plantear cuestiones exclusivas debido a su naturaleza única. Algunas áreas tecnológicas, como Inteligencia Artificial (IA) y Ciberseguridad son relativamente nuevos. La FDA ha colaborado con la industria para desarrollar documentos de orientación en estas áreas.
En estos casos, se recomienda encarecidamente una reunión previa con la FDA antes de la presentación del 510(k). Remita a la Documento guía de la FDA si las empresas necesitan información y reunirse con la FDA antes de la presentación del 510(k).
Conclusión:
Estos eran los puntos importantes de la lista de comprobación de una presentación 510(k). Las cartas de denegación de aceptación (RTA) de la FDA podrían evitarse fácilmente mediante una revisión cuidadosa de la presentación y siguiendo los documentos de orientación de la FDA.
La solicitud de información adicional como parte del proceso de revisión de fondo podría reducirse mediante la redacción de un 510(k) claro y conciso. Esto suele ser una combinación de ciencia, arte y experiencia. Se recomienda encarecidamente contar con la ayuda de expertos Consultores de asuntos reglamentarios en la redacción del 510(k) para evitar costosos errores siga esta lista de comprobación para la presentación del 510(k). Manténgase al día en cuanto a los reglamentos, normas y documentos de orientación aplicables para aumentar las posibilidades de éxito de la presentación.
Necesidad de consultar a un Experto en presentaciones a la FDA? Trabaje con redactores de normativas experimentados, expertos de la industria de dispositivos médicos y Consultores 510k que han ayudado a las empresas de dispositivos médicos a preparar los documentos reglamentarios para obtener la autorización de la FDA.
Kolabtree helps businesses worldwide hire freelance scientists and industry experts on demand. Our freelancers have helped companies publish research papers, develop products, analyze data, and more. It only takes a minute to tell us what you need done and get quotes from experts for free.

Unlock Corporate Benefits
• Secure Payment Assistance
• Onboarding Support
• Dedicated Account Manager
Sign up with your professional email to avail special advances offered against purchase orders, seamless multi-channel payments, and extended support for agreements.


