

Nare Simonyan, freelance regulatory affairs specialist di Kolabtree, fornisce una guida completa a un dossier normativo e al suo formato.
Introduzione: Cos'è un dossier regolamentare?
Regulatory dossier is a package of documents, which may include all required information regarding newly developed drug products and/or generics, which is required by EU and US regulatory authorities for granting marketing authorization approvals. The main information that is included in the package is administrative information, data related to the quality, safety and efficacy of drug product, which can be submitted by CTD (Common Technical Document) format both paper and electronic version. Since the information submitted in paper format was enormous, agencies are now encouraging applications to be submitted in eCTD format.
CTD (documento tecnico comune)
Nel 2003, i membri dell'ICH (International Council of Harmonization) si sono accordati per riunire tutte le informazioni di qualità, sicurezza ed efficacia in un formato comune che si chiamava CTD. Il CTD è un formato/struttura per i moduli da 1 a 5 della NDA (New Drug Application), MAA (Marketing Authorization Application), e delle applicazioni globali di medicinali. Il modulo 1 contiene informazioni amministrative regionali che sono diverse per ogni paese. I moduli 2, 3, 4 e 5 sono comuni a tutte le regioni. La struttura CTD si applica sia alle applicazioni sperimentali che a quelle commerciali (IND (Investigational New Drug) e NDA (USA); IMPD (Investigational Medicinal Product Dossier) e MAA (EU), e applicazioni globali).
The CTD was primarily used for new marketing applications such as NDA, BLA (Biologics License Application), MAA, NDS (New Drug Substance), JNDA (Japanese New Drug Application), etc. With additional guidance and guidelines from the FDA and EMA, the CTD is now required for all applications, including those for studi clinici—IMPD and INDs. All Drug Master Files (DMF) and Active Substance Master Files (ASMF) must follow the structure of the CTD. The process of submitting regulatory dossier is regulated by Code of Federal Regulations (CFR) (Law in US) and Directives (Law in EU).
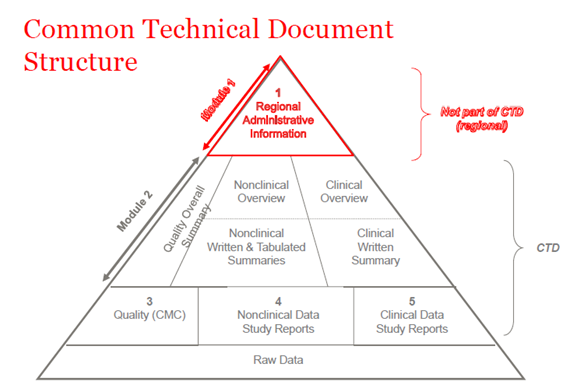

Figura 1. La struttura del CTD
CTD-Modulo 1
Specifiche per US Module 1: Informazioni amministrative e informazioni sulla prescrizione
La sezione e-CTD Modulo 1 contiene documenti amministrativi e di etichettatura. Tutte le domande e le relative presentazioni hanno la stessa struttura organizzativa per i documenti del Modulo 1.
Si prega di vedere di seguito alcune specifiche sui documenti da fornire all'interno del Modulo 1:
Lettera di presentazione (sezione 1.2)
Le lettere di presentazione contengono informazioni pertinenti che aiutano la comunicazione nel processo di revisione. Si raccomanda di includere le seguenti informazioni nella lettera di accompagnamento (si prega di fare riferimento a "Documento tecnico comune elettronico (eCTD) v4.0 GUIDA ALLA CONFORMITÀ TECNICA")
- Descrizione normativa della presentazione, comprese le informazioni normative appropriate, e qualsiasi collegamento ipertestuale desiderato alle informazioni presentate
- Descrizione tecnica della presentazione, compresa la dimensione approssimativa della presentazione (ad esempio, 2 gigabyte)
- Dichiarazione che l'invio è privo di virus, con una descrizione del software (nome, versione e società) che è stato utilizzato per controllare i file per i virus
- Un punto di contatto normativo e tecnico per la presentazione, compreso l'indirizzo e-mail.
Certificazione delle copie sul campo (sezione 1.3)
La certificazione della copia di campo deve essere inclusa nell'eCTD per le domande di commercializzazione. Può essere una lettera all'ufficio distrettuale che notifica che la presentazione eCTD sarà presentata alla FDA. La lettera dovrebbe includere:
- Droga e numero di applicazione
- Centro e divisione FDA
- La domanda è in formato eCTD.
Riferimenti (sezione 1.4)
I riferimenti possono contenere le seguenti sottosezioni (possono essere richiesti anche contenuti aggiuntivi)
- Lettere di autorizzazione (LOA)
- Riferimento incrociato di informazioni precedentemente inviate che non sono in formato eCTD
Modifiche delle informazioni (sezione 1.11)
Questo può essere usato per la presentazione di risposte a richieste di informazioni (IR), quando le informazioni presentate non rientrano in nessuna voce del Modulo 2, 3, 4 o 5. Se la risposta all'IR ha un impatto sui documenti presentati nei Moduli 2 - 5, i documenti nuovi o sostitutivi devono essere presentati nella posizione appropriata del Modulo 2 - 5 e citati nel documento della Sezione 1.11.
Rapporti annuali di marketing (sezione 1.13)
Per ogni studio o prova descritta nei file dei requisiti/impegni post marketing deve essere incluso un segnalibro.
Etichettatura (sezione 1.14)
La sezione 1.14 descrive come fornire documenti di etichettatura specifici. Può includere l'etichettatura della bozza, l'etichettatura finale, l'etichettatura dei farmaci elencati, l'etichettatura dei farmaci in fase di sperimentazione, l'etichettatura estera e l'etichettatura del prodotto per le presentazioni 2253*. Le informazioni fornite possono contenere la storia, il contenuto e i campioni dell'etichettatura.
Modulo FDA 2253*: Trasmissione di pubblicità ed etichettatura promozionale per farmaci e biologici per uso umano
Pubblicità e materiale di etichettatura promozionale (Sezione 1.15)
Le pubblicità e i materiali promozionali in etichetta sono limitati negli Stati Uniti, e dovrebbero riflettersi ai requisiti menzionati nella guida della FDA Fornire le presentazioni regolamentari in formato elettronico e non elettronico - Etichettatura promozionale e materiale pubblicitario per i farmaci da prescrizione per uso umano.
Per i materiali promozionali presentati come parte dei requisiti di segnalazione post-commercializzazione, si suggerisce di fornire collegamenti ipertestuali a riferimenti o etichettature. I riferimenti migliorano l'efficienza di una revisione.
Strategia di valutazione e mitigazione del rischio (REMS) (sezione 1.16)
Il supplemento REMS è destinato a proporre un nuovo REMS o modifiche (maggiori e/o minori) ad un REMS approvato. Nel modulo FDA appropriato deve essere selezionato il tipo di supplemento REMS. Le valutazioni REMS, le revisioni REMS e le corrispondenze REMS non sono supplementi. In questo caso tipo di presentazione deve essere selezionato "altro" quando si compila il modulo FDA.
Specifiche per l'Unione Europea (UE) Modulo 1: Informazioni amministrative e informazioni per la prescrizione
Lettera di presentazione (sezione 1.0)
Per ogni domanda deve essere fornita una lettera di accompagnamento. I documenti "Note ai revisori" possono essere inclusi come appendice alla lettera di accompagnamento, nel caso in cui sia necessario fornire ulteriori informazioni per facilitare la navigazione.
Indice completo (sezione 1.1)
Per ogni tipo di domanda deve essere fornito un indice completo, che può contenere tutte le sezioni del modulo che sono state presentate come parte della domanda in questione. Nel caso di nuove domande, tutte le sezioni devono essere trattate.
Modulo di domanda (sezione 1.2)
A seconda del tipo di presentazione, il relativo modulo di domanda dovrebbe essere incluso nel dossier regolamentare.
I diversi moduli di domanda sono disponibili sul sito web della Commissione europea /
DG Impresa:
- Nuove applicazioni e applicazioni di estensione http://ec.europa.eu/enterprise/pharmaceuticals/eudralex/homev2.htm#2b
- Applicazioni di variazione
http://ec.europa.eu/enterprise/pharmaceuticals/eudralex/homev2.htm#2c
- Domande di rinnovo
http://ec.europa.eu/enterprise/pharmaceuticals/eudralex/homev2.htm#2c
Informazioni sul prodotto (sezione 1.3)
La sezione 1.3 contiene informazioni riguardanti
- Riassunto delle caratteristiche del prodotto (SPC), etichettatura e foglietto illustrativo (PIL) (Sezione 1.3.1)
- Mock-up (Sezione 1.3.2)
- Campione (Sezione 1.3.3)
- Consultazione con i gruppi di pazienti target (Sezione 1.3.4)
- Informazioni sul prodotto già approvato negli Stati membri (Sezione 1.3.5) (dove applicabile)
- Braille (Sezione 1.3.6)
Informazioni sugli esperti (sezione 1.4)
In questa sezione è necessario includere le informazioni relative agli esperti che forniscono relazioni dettagliate dei documenti e delle informazioni che costituiscono i moduli 3, 4 e 5 in conformità con l'articolo 12 della direttiva 2001/83 / CE. Per la parte aggiuntiva di questa sezione può essere utilizzato un rapporto di esperti firmato per le diverse parti scientifiche del dossier. I requisiti firmati relazioni di esperti presentati di seguito:
- Il Quality Overall Summary, Non-clinical Overview / Summary e Clinical Overview / Summary nel Modulo 2,
- Una dichiarazione firmata dagli esperti del Modulo 1.4.
- Una breve informazione sul background educativo, la formazione e l'esperienza professionale nel Modulo 1.4.
Le dichiarazioni degli esperti devono essere fornite per le domande post-autorizzazione.
Nei casi in cui i titolari dell'autorizzazione all'immissione in commercio desiderano distinguere tale dichiarazione da qualsiasi
dichiarazioni precedenti, il numero di procedura pertinente dello stato membro/SEE di riferimento
può essere incluso sopra.
Le sottosezioni delle informazioni sugli esperti sono presentate qui sotto:
- Qualità (sezione 1.4.1)
- Non clinico (1.4.2)
- Clinica (Sezione1.4.3)
Per ulteriori informazioni e modelli pertinenti si prega di fare riferimento all'articolo 12 e in conformità con l'allegato I, parte I 1.4 della direttiva 2001/83/CE.
Requisiti specifici per diversi tipi di applicazioni (sezione 1.5)
Informazioni per applicazioni bibliografiche (sezione 1.5.1)
In base all'articolo 10a della direttiva 2001/83/CE, i richiedenti devono fornire un documento conciso che riassuma i motivi e le prove utilizzati per dimostrare che il componente o i componenti del medicinale hanno un uso ben noto, tenendo conto del livello accettabile di sicurezza ed efficacia, come indicato nella parte II.1 dell'allegato I della direttiva 2001/83/CE.
Informazioni per applicazioni generiche, "ibride" o biosimilari (sezione 1.5.2)
Sulla base dell'articolo 10(1), 10(3) o 10(4) della direttiva 2001/83/CE, i richiedenti devono fornire qui un documento conciso che riassuma le motivazioni e le prove utilizzate per dimostrare il tipo di medicinale per il quale viene presentata la domanda. I medicinali possono essere generici, prodotti ibridi e biosimilari.
(Esteso) Dati / Esclusività di mercato (Sezione 1.5.3)
La sezione 1.5.3 è necessaria quando il titolare/richiedente dell'autorizzazione all'immissione in commercio desidera rivendicare dati/esclusività di mercato (aggiuntivi) mentre fa domanda per una nuova indicazione o un cambiamento di classificazione. In questo caso devono essere prese in considerazione le disposizioni e i requisiti legali pertinenti.
Circostanze eccezionali (sezione 1.5.4)
Secondo l'articolo 22 della direttiva 2001/83/CE e l'articolo 14, paragrafo 7, del regolamento (CE) n. 726/2004, un'autorizzazione può essere concessa in caso di circostanze eccezionali, quando il richiedente introduce procedure specifiche, in particolare per quanto riguarda la sicurezza del medicinale, la notifica alle autorità competenti di qualsiasi incidente relativo al suo uso. Solo per eccezioni oggettive e verificabili può essere concessa un'autorizzazione.
Autorizzazione all'immissione in commercio condizionata (sezione 1.5.5)
La sezione citata è applicabile alla procedura centralizzata. I riferimenti per questa sezione sono l'articolo 14, paragrafo 7, del regolamento (CE) n. 726/2004 e "Linee guida sull'applicazione scientifica e le modalità pratiche dell'autorizzazione all'immissione in commercio condizionata".
Valutazione del rischio ambientale (sezione 1.6)
In conformità con l'articolo 8 (ca) e (g) della direttiva 2001/83/CE, qualsiasi rischio potenziale del medicinale per l'ambiente deve essere considerato dal richiedente durante la richiesta di approvazione dell'autorizzazione alla commercializzazione. I requisiti della direttiva riguardano l'uso, la conservazione e lo smaltimento dei medicinali e non sono applicabili alla sintesi o alla fabbricazione del prodotto. Le domande di autorizzazione all'immissione in commercio di medicinali che non contengono OGM (sezione 1.6.1 Non OGM) e che contengono OGM (1.6.2 OGM) devono includere un'indicazione di tutti i rischi potenziali nel modulo 1 del dossier normativo.
Per ulteriori informazioni si prega di fare riferimento a "Linee guida sulla valutazione del rischio ambientale per i medicinali per uso umano"
Informazioni relative all'esclusività del mercato orfano (sezione 1.7)
Questa sezione è applicabile solo per i farmaci orfani. Le informazioni necessarie sui dettagli e la procedura sono presenti in "Linea guida della Commissione europea sugli aspetti dell'applicazione dell'articolo 8 del regolamento (CE) n. 141/2000: Valutazione della somiglianza e/o della superiorità clinica dei medicinali orfani nella valutazione delle domande di autorizzazione all'immissione in commercio e delle variazioni."
Se la sezione 1.7.1 Similarità e la sezione 1.7.2 Esclusività di mercato sono applicabili, se il medicinale orfano è stato autorizzato per l'affezione che copre l'indicazione terapeutica proposta oggetto della domanda e se è in vigore un periodo di esclusiva di mercato, il richiedente deve fornire una relazione critica che esamini la possibile somiglianza con il medicinale orfano autorizzato e che concluda sulla somiglianza o sulla "non" somiglianza (1.7.1). Se il medicinale oggetto della domanda di autorizzazione all'immissione in commercio è ritenuto "simile" a un medicinale orfano coperto dalle disposizioni di esclusiva di mercato di cui sopra, il richiedente deve inoltre fornire la giustificazione che una delle deroghe previste all'articolo 8.3, lettere da a) a c), del regolamento (CE) n. 141/2000(1.7.2).
Information relating to Farmacovigilanza (Sezione 1.8)
Sistema di farmacovigilanza (sezione 1.8.1)
La farmacovigilanza è la scienza e le attività legate al rilevamento, alla valutazione, alla comprensione e alla prevenzione degli effetti avversi o di qualsiasi altro problema legato ai farmaci.
Questo vale per tutto il ciclo di vita del farmaco, sia nella fase di pre-approvazione che in quella post-approvazione. Il sistema di farmacovigilanza è una sezione molto importante per l'applicazione dell'autorizzazione alla commercializzazione.
Deve essere fornita una descrizione dettagliata del sistema di farmacovigilanza secondo l'articolo 8 (ia) della direttiva 2001/83/CE. La descrizione deve includere la prova che il richiedente ha i servizi di una persona qualificata responsabile della farmacovigilanza.
La descrizione del sistema di farmacovigilanza del titolare dell'autorizzazione all'immissione in commercio deve seguire i requisiti e il formato dettagliati nel volume 9A di EudraLex.
Sistema di gestione dei rischi (sezione 1.8.2)
Detailed description of risk-management system must be provided according to Article 8 (ia) of Directive 2001/83/EC. The detailed description of a risk management system should be provided in the form of an EU Risk Management Plan (EU-RMP), as outlined in Volume 9A of EudraLex.
Informazioni relative agli studi clinici (sezione 1.9)
La sezione del 1.9 deve essere preparata secondo l'articolo 8 (ib) della direttiva 2001/83/CE per la relazione che le sperimentazioni cliniche effettuate al di fuori dell'Unione europea soddisfano i requisiti etici della direttiva 2001/20/CE deve essere fornita, se applicabile.
Questa sezione deve essere fornita per tutte le nuove domande (comprese le domande di estensione), e altre procedure normative post-autorizzazione pertinenti (ad esempio, le variazioni) per le quali la sperimentazione clinica ha presentato rapporti.
Informazioni relative alla pediatria (sezione 1.10)
Conformemente agli articoli 7, 8 e 30 del regolamento (CE) n. 1901/2006 ("regolamento pediatrico") e all'articolo 23 del regolamento (CE) n. 1901/2006 ("regolamento pediatrico") questa sezione è obbligatoria:
- Per tutte le nuove domande*, per un medicinale che non è autorizzato nel SEE
- Per le domande* di nuove indicazioni, nuove forme farmaceutiche e nuove vie di somministrazione, per i medicinali autorizzati che sono protetti da un certificato di protezione complementare o da un brevetto che si qualifica per il rilascio di un tale certificato.
- Per le domande di autorizzazione alla commercializzazione per uso pediatrico (PUMA)
*fatta eccezione per le applicazioni generiche, ibride, biosimilari e di uso consolidato e per i medicinali tradizionali a base di erbe o omeopatici
Per il Modulo 1 può essere fornito:
Risposte alle domande nei casi in cui si consiglia ai richiedenti di includere in questa sezione un documento che elenchi le domande con la corrispondente risposta testuale narrativa per ogni domanda, e quando le risposte contengono anche dati/documenti nuovi o aggiornati relativi ai Moduli 3, 4 e/o 5. Tali dati/documenti dovrebbero essere inseriti nelle sezioni pertinenti di tali moduli.
Dati aggiuntivi. Questa sezione è richiesta in base alla procedura di autorizzazione. Potrebbe essere necessario fornire ulteriori dati come parte di una domanda nazionale, decentralizzata o di riconoscimento reciproco. Se tali dati si riferiscono ai Moduli 2, 3, 4 e/o 5, i documenti dovrebbero essere inseriti anche nelle sezioni pertinenti di tali Moduli. I requisiti specifici degli Stati membri per i dati aggiuntivi possono essere trovati sul sito web del Commissione europea.
CTD-Modulo 2
L'introduzione generale al medicinale fornita nella sezione Modulo 2 del dossier CTD, che è armonizzato per tutte le regioni (Il Consiglio internazionale per l'armonizzazione dei requisiti tecnici dei prodotti farmaceutici per uso umano (ICH)). Questo modulo è presentato da documenti riassuntivi per ogni modulo successivo: dati di qualità, rapporti di studi non clinici e clinici.
CTD-Modulo 3
Module 3 was identified by ICH as the Quality Module. Thus, “Quality” became the global term for CMC (chimica, manufacturing and controls). Il CMC non deve essere confuso con gli elementi del controllo di qualità, della garanzia di qualità, delle SOP (procedure operative standard), dei documenti aziendali interni (specifiche, registri dei lotti, ecc.)
La sezione del modulo 3 è anche armonizzata per tutte le regioni con la fornitura di informazioni chimico-farmaceutiche e biologiche per i principi attivi chimici e i medicinali biologici.
Durante tutto lo sviluppo e dopo che un prodotto è approvato dalle autorità sanitarie, gli aspetti di chimica, produzione e controllo (CMC) continuano ad evolversi e a cambiare.
Qualità farmaceutica = CMC
CHIMICA
Scoperta di nuove entità chimiche/molecole, purificazione di principi attivi (sostanza farmaceutica), test analitici di materie prime e prodotti finiti.
MANIFATTURAZIONE
Produzione su scala di laboratorio di sostanze e prodotti farmaceutici, produzione di forniture cliniche per studi clinici, scalare fino alla dimensione del lotto commerciale, prodotto commerciale.
E CONTROLLI
Sviluppare specifiche/controlli appropriati per la sostanza e il prodotto farmaceutico per garantire sicurezza, efficacia e qualità.
Tutte le regioni richiedono il dossier CMC/qualità farmaceutica
- Avviare e condurre studi clinici,
- Per presentare un nuovo dossier di autorizzazione alla commercializzazione (MAA, NDA, BLA, ecc.)
- Soddisfare i requisiti normativi per la gestione del ciclo di vita e le modifiche post approvazione del prodotto
Questo modulo include l'intero pacchetto di documenti riguardanti la sostanza farmaceutica (DS) e il prodotto farmaceutico (DP).
Sostanza stupefacente (DS)
La sezione della sostanza farmaceutica è presentata dalla sua descrizione, comprese le caratteristiche fisiche, chimiche o biologiche, il nome e l'indirizzo del produttore DS, i limiti accettabili e i metodi analitici utilizzati per assicurare l'identità, la forza, la qualità e la purezza della sostanza farmaceutica.
Sono anche incluse informazioni a sostegno della stabilità della sostanza farmaceutica durante gli studi tossicologici e lo studio clinico proposto.
PRODOTTO FARMACEUTICO (DP)
Un elenco di tutti i componenti, che possono includere alternative ragionevoli per i composti inattivi, utilizzati nella fabbricazione del prodotto farmaceutico, compresi sia i componenti destinati a comparire nel prodotto farmaceutico che quelli che possono non comparire, ma che sono utilizzati nel processo di fabbricazione sono descritti in questa sezione.
Di seguito sono elencate le informazioni chiave del prodotto farmaceutico che devono essere incluse nel dossier normativo:
- La composizione quantitativa del prodotto farmaceutico (sperimentale o commerciale), compresa qualsiasi variazione ragionevole.
- Il nome e l'indirizzo del produttore del prodotto farmaceutico.
- Descrizione delle procedure di fabbricazione e imballaggio.
- I limiti accettabili e i metodi analitici utilizzati per assicurare l'identità, la forza, la qualità e la purezza del prodotto farmaceutico.
CTD-Modulo 4
Rapporti di studi non clinici
La sezione pertinente la posizione appropriata per i dati sui singoli animali è nel rapporto di studio nel documento tecnico comune per le applicazioni che saranno presentate alle autorità regolatorie. Il modulo 4 è armonizzato per gli USA e l'UE sulla base dei principi ICH e contiene tutte le sezioni e sottosezioni necessarie per i rapporti di studio. Riferimenti alle linee guida ICH Documento Tecnico Comune (CTD).
CTD-Modulo 5
Rapporti di studi clinici
La sezione del modulo 5 riguarda la struttura e il contenuto dei rapporti degli studi clinici. Questa parte del CTD presenta i rapporti di studio umano/clinico, altri dati clinici e riferimenti all'interno di un documento tecnico comune (CTD) per la registrazione di un prodotto farmaceutico per uso umano. Questi elementi dovrebbero facilitare la preparazione e la revisione di una domanda di commercializzazione. Il modulo 5 è armonizzato per gli USA e l'UE sulla base dei principi ICH e contiene tutte le sezioni e sottosezioni necessarie per i rapporti di studio. Riferimenti alle linee guida UE Documento tecnico comune (CTD).
Hai bisogno di aiuto per preparare un dossier regolamentare? Guarda e consulta scrittori freelance di normative su Kolabtree.
Riferimenti aggiuntivi
- https://www.ich.org/page/ctd
- https://www.fda.gov/media/128163/download
- https://www.fda.gov/media/135573/download
- https://ec.europa.eu/health/sites/default/files/files/eudralex/vol-2/b/update_200805/ctd_05-2008_en.pdf
- https://ec.europa.eu/health/sites/default/files/files/eudralex/vol-1/dir_2001_83_consol_2012/dir_2001_83_cons_2012_en.pdf
- https://eur-lex.europa.eu/legal-content/EN/TXT/PDF/?uri=CELEX:02001L0083-20121116&from=DE
- https://eur-lex.europa.eu/legal-content/EN/TXT/PDF/?uri=CELEX:02004R0726-20130605&from=EN
- https://ec.europa.eu/health/sites/default/files/files/eudralex/vol-1/reg_2000_141_cons-2009-07/reg_2000_141_cons-2009-07_en.pdf
- https://www.ema.europa.eu/en/human-regulatory/research-development/clinical-trials-human-medicines
- https://eur-lex.europa.eu/legal-content/EN/TXT/?uri=celex%3A32006R1901
- https://clinicaltrials.gov/ct2/home
Kolabtree helps businesses worldwide hire freelance scientists and industry experts on demand. Our freelancers have helped companies publish research papers, develop products, analyze data, and more. It only takes a minute to tell us what you need done and get quotes from experts for free.


Unlock Corporate Benefits
• Secure Payment Assistance
• Onboarding Support
• Dedicated Account Manager
Sign up with your professional email to avail special advances offered against purchase orders, seamless multi-channel payments, and extended support for agreements.