

Nare Simonyan, freelance regulatory affairs specialist bei Kolabtree, bietet einen umfassenden Leitfaden für ein Regulierungsdossier und dessen Format.
Einleitung: Was ist ein Regulierungsdossier?
Das Zulassungsdossier ist ein Paket von Dokumenten, das alle erforderlichen Informationen zu neu entwickelten Arzneimitteln und/oder Generika enthalten kann, die von den Zulassungsbehörden in der EU und den USA für die Erteilung der Marktzulassung verlangt werden. Die wichtigsten Informationen, die in dem Paket enthalten sind, sind administrative Informationen, Daten in Bezug auf die Qualität, Sicherheit und Wirksamkeit des Arzneimittels, die im CTD-Format (Common Technical Document) sowohl in Papierform als auch in elektronischer Form eingereicht werden können. Da die in Papierform eingereichten Informationen sehr umfangreich waren, fordern die Behörden nun dazu auf, die Anträge im eCTD-Format einzureichen.
CTD (Gemeinsames Technisches Dokument)
Im Jahr 2003 haben sich die Mitglieder der ICH (International Council of Harmonization) darauf geeinigt, alle Informationen über Qualität, Sicherheit und Wirksamkeit in einem gemeinsamen Format zusammenzufassen, das früher CTD genannt wurde. Das CTD ist ein Format bzw. eine Struktur für die Module 1 bis 5 der NDA (New Drug Application), MAA (Marketing Authorization Application) und der globalen medizinischen Anträge. Modul 1 enthält administrative regionale Informationen, die für jedes Land unterschiedlich sind. Die Module 2, 3, 4 und 5 sind für alle Regionen gleich. Die CTD-Struktur gilt sowohl für Prüfanträge als auch für kommerzielle Anträge (IND (Investigational New Drug) und NDA (USA); IMPD (Investigational Medicinal Product Dossier) und MAA (EU) sowie für globale Anträge).
The CTD was primarily used for new marketing applications such as NDA, BLA (Biologics License Application), MAA, NDS (New Drug Substance), JNDA (Japanese New Drug Application), etc. With additional guidance and guidelines from the FDA and EMA, the CTD is now required for all applications, including those for klinische Studien—IMPD and INDs. All Drug Master Files (DMF) and Active Substance Master Files (ASMF) must follow the structure of the CTD. The process of submitting regulatory dossier is regulated by Code of Federal Regulations (CFR) (Law in US) and Directives (Law in EU).
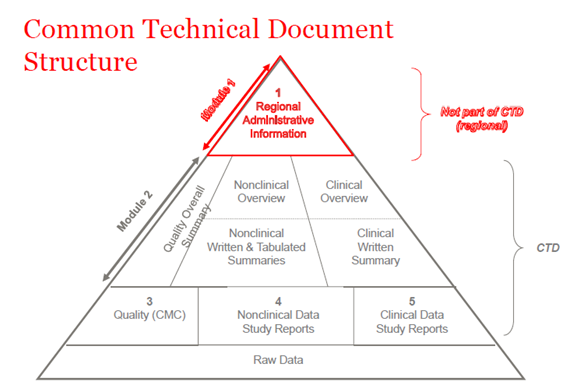

Abbildung 1. Die Struktur von CTD
CTD-Baustein 1
Spezifikationen für US-Modul 1: Administrative Informationen und Verschreibungsinformationen
Der Abschnitt e-CTD Modul 1 enthält administrative und kennzeichnungsbezogene Dokumente. Alle Anträge und damit zusammenhängenden Einreichungen haben die gleiche Organisationsstruktur für Dokumente des Moduls 1.
Nachstehend finden Sie einige Angaben zu den Dokumenten, die im Rahmen von Modul 1 vorzulegen sind:
Anschreiben (Abschnitt 1.2)
Anschreiben enthalten sachdienliche Informationen, die die Kommunikation im Rahmen des Überprüfungsverfahrens erleichtern. Die folgenden Informationen sollten in das Anschreiben aufgenommen werden (siehe "Elektronisches Gemeinsames Technisches Dokument (eCTD) v4.0 TECHNICAL CONFORMANCE GUIDE")
- Regulatorische Beschreibung der Einreichung, einschließlich der entsprechenden regulatorischen Informationen, und alle gewünschten Hyperlinks zu den eingereichten Informationen
- Technische Beschreibung des Beitrags, einschließlich der ungefähren Größe des Beitrags (z. B. 2 Gigabyte)
- Erklärung, dass die Einsendung virenfrei ist, mit einer Beschreibung der Software (Name, Version und Unternehmen), die zur Überprüfung der Dateien auf Viren verwendet wurde
- Eine Kontaktstelle für regulatorische und technische Fragen für die Einreichung, einschließlich einer E-Mail-Adresse.
Zertifizierung der Feldkopie (Abschnitt 1.3)
Die Zertifizierung der Feldkopie sollte im eCTD für Zulassungsanträge enthalten sein. Dies kann ein Brief an das Bezirksamt sein, in dem mitgeteilt wird, dass die eCTD-Einreichung bei der FDA eingereicht wird. Das Schreiben sollte Folgendes enthalten:
- Medikament und Antragsnummer
- FDA-Zentrum und -Abteilung
- Die Bewerbung erfolgt im eCTD-Format.
Referenzen (Abschnitt 1.4)
Die Referenzen können folgende Unterabschnitte enthalten (es können auch zusätzliche Inhalte verlangt werden)
- Bevollmächtigungsschreiben (LOAs)
- Querverweise zu früher eingereichten Informationen, die nicht im eCTD-Format vorliegen
Änderungen der Informationen (Abschnitt 1.11)
Dies kann für die Übermittlung von Antworten auf Informationsanfragen (IR) verwendet werden, wenn die zu übermittelnden Informationen nicht in eine der Rubriken der Module 2, 3, 4 oder 5 passen. Wenn sich die Antwort auf eine Informationsanfrage auf Dokumente auswirkt, die in den Modulen 2 bis 5 eingereicht wurden, sollten die neuen oder zu ersetzenden Dokumente an der entsprechenden Stelle in den Modulen 2 bis 5 eingereicht werden und in dem Dokument aus Abschnitt 1.11 referenziert werden.
Marketing-Jahresberichte (Abschnitt 1.13)
Für jede Studie oder Prüfung, die in den Unterlagen zu den Anforderungen/Verpflichtungen nach dem Inverkehrbringen beschrieben ist, sollte ein Lesezeichen enthalten sein.
Kennzeichnung (Abschnitt 1.14)
In Abschnitt 1.14 wird beschrieben, wie spezifische Kennzeichnungsdokumente bereitzustellen sind. Dies kann Kennzeichnungsentwürfe, endgültige Kennzeichnungen, Kennzeichnungen für gelistete Arzneimittel, Kennzeichnungen für Prüfpräparate, ausländische Kennzeichnungen und Produktkennzeichnungen für 2253 Einreichungen* umfassen. Die bereitgestellten Informationen können die Geschichte, den Inhalt und Muster der Kennzeichnung enthalten.
Formular FDA 2253*: Übermittlung von Anzeigen und Werbeaufschriften für Humanarzneimittel und biologische Arzneimittel
Werbung und Kennzeichnungsmaterial zu Werbezwecken (Abschnitt 1.15)
Werbung und Werbematerialien sind in den USA eingeschränkt, sie sollten den Anforderungen der FDA-Leitlinien entsprechen. Einreichung von Zulassungsanträgen in elektronischem und nicht-elektronischem Format - Werbeetikettierung und Werbematerial für verschreibungspflichtige Humanarzneimittel.
Bei Werbematerialien, die im Rahmen der Berichtspflicht nach dem Inverkehrbringen eingereicht werden, sollten Hypertext-Links zu Referenzen oder Kennzeichnungen angegeben werden. Referenzen verbessern die Effizienz einer Überprüfung.
Strategie zur Risikobewertung und -minderung (REMS) (Abschnitt 1.16)
Ein REMS-Supplement dient dazu, ein neues REMS oder Änderungen (größere und/oder kleinere) an einem genehmigten REMS vorzuschlagen. Im entsprechenden FDA-Formular muss der Typ REMS-Supplement ausgewählt werden. REMS-Bewertungen, REMS-Überarbeitungen und REMS-Korrespondenzen sind keine Ergänzungen. In diesem Fall Abgabeart sollte beim Ausfüllen des FDA-Formulars die Option "Sonstige" gewählt werden.
Spezifikationen für die Europäische Union (EU) Modul 1: Administrative Informationen und Verschreibungsinformationen
Anschreiben (Abschnitt 1.0)
Für jeden Antrag sollte ein Anschreiben erstellt werden. Dem Anschreiben können Dokumente mit "Hinweisen für die Prüfer" beigefügt werden, falls zur Erleichterung der Navigation weitere Informationen benötigt werden.
Umfassendes Inhaltsverzeichnis (Abschnitt 1.1)
Für jede Art von Antrag sollte ein umfassendes Inhaltsverzeichnis erstellt werden, das alle Modulabschnitte enthalten kann, die als Teil des betreffenden Antrags eingereicht wurden. Bei neuen Anträgen sollten alle Abschnitte behandelt werden.
Antragsformular (Abschnitt 1.2)
Je nach Art der Einreichung sollte das entsprechende Antragsformular in das Regulierungsdossier aufgenommen werden.
Die verschiedenen Antragsformulare finden Sie auf der Website der Europäischen Kommission /
GD Unternehmen:
- Neue Anträge und Erweiterungsanträge http://ec.europa.eu/enterprise/pharmaceuticals/eudralex/homev2.htm#2b
- Anträge auf Änderung
http://ec.europa.eu/enterprise/pharmaceuticals/eudralex/homev2.htm#2c
- Verlängerungsanträge
http://ec.europa.eu/enterprise/pharmaceuticals/eudralex/homev2.htm#2c
Produktinformation (Abschnitt 1.3)
Abschnitt 1.3 enthält Informationen über
- Zusammenfassung der Produktmerkmale (SPC), Etikettierung und Packungsbeilage (PIL) (Abschnitt 1.3.1)
- Attrappe (Abschnitt 1.3.2)
- Probe (Abschnitt 1.3.3)
- Konsultation mit Patienten-Zielgruppen (Abschnitt 1.3.4)
- In den Mitgliedstaaten bereits genehmigte Produktinformation (Abschnitt 1.3.5) (falls zutreffend)
- Blindenschrift (Abschnitt 1.3.6)
Informationen über die Experten (Abschnitt 1.4)
In diesem Abschnitt sind Angaben zu den Sachverständigen zu machen, die ausführliche Berichte über die Dokumente und Informationen erstellen, die gemäß Artikel 12 der Richtlinie 2001/83/EG die Module 3, 4 und 5 bilden. Als zusätzlicher Teil dieses Abschnitts kann ein unterzeichneter Expertenbericht für die verschiedenen wissenschaftlichen Teile des Dossiers verwendet werden. Die Anforderungen an die unterzeichneten Sachverständigenberichte sind unten aufgeführt:
- Die Qualitäts-Gesamtzusammenfassung, die nicht-klinische Übersicht/Zusammenfassung und die klinische Übersicht/Zusammenfassung in Modul 2,
- Eine von den Experten in Modul 1.4 unterzeichnete Erklärung.
- Eine kurze Information über den Bildungshintergrund, die Ausbildung und die Berufserfahrung in Modul 1.4.
Bei Anträgen auf Nachzulassung ist/sind die entsprechende(n) Expertenerklärung(en) vorzulegen.
In den Fällen, in denen die Zulassungsinhaber eine solche Erklärung von einer
frühere Anmeldungen, die entsprechende Verfahrensnummer des Referenzmitgliedstaates/des EWR
kann zusätzlich aufgenommen werden.
Nachfolgend werden Unterabschnitte der Informationen über die Experten vorgestellt:
- Qualität (Abschnitt 1.4.1)
- Nichtklinisch (1.4.2)
- Klinisch (Abschnitt 1.4.3)
Weitere Informationen und einschlägige Vorlagen finden Sie in Artikel 12 und in Übereinstimmung mit Anhang I, Teil I 1.4 der Richtlinie 2001/83/EG.
Spezifische Anforderungen für verschiedene Arten von Anwendungen (Abschnitt 1.5)
Informationen für bibliografische Anwendungen (Abschnitt 1.5.1)
Auf der Grundlage von Artikel 10a der Richtlinie 2001/83/EG sollten die Antragsteller ein kurzes Dokument vorlegen, in dem die Gründe und Beweise für den Nachweis zusammengefasst sind, dass der/die Bestandteil(e) des Arzneimittels allgemein gebräuchlich ist/sind, wobei ein annehmbarer Grad an Sicherheit und Wirksamkeit gemäß Anhang I Teil II.1 der Richtlinie 2001/83/EG zu berücksichtigen ist.
Informationen für Anträge auf Generika, 'hybride' Arzneimittel oder Bio-Similars (Abschnitt 1.5.2)
Auf der Grundlage von Artikel 10 Absatz 1, 3 oder 4 der Richtlinie 2001/83/EG sollten die Antragsteller hier ein kurzes Dokument einreichen, in dem die Gründe und Nachweise für die Art des Arzneimittels, für das ein Antrag gestellt wird, zusammengefasst sind. Bei den Arzneimitteln kann es sich um Generika, Hybridprodukte und Biosimilars handeln.
(Erweiterte) Daten-/Marktexklusivität (Abschnitt 1.5.3)
Abschnitt 1.5.3 ist erforderlich, wenn der Zulassungsinhaber/Antragsteller (zusätzliche) Daten/Marktexklusivität beanspruchen möchte, während er eine neue Indikation oder eine Änderung der Einstufung beantragt. In diesem Fall müssen die einschlägigen gesetzlichen Bestimmungen und Anforderungen berücksichtigt werden.
Außergewöhnliche Umstände (Abschnitt 1.5.4)
Gemäß Artikel 22 der Richtlinie 2001/83/EG und Artikel 14 Absatz 7 der Verordnung (EG) Nr. 726/2004 kann eine Genehmigung erteilt werden, wenn außergewöhnliche Umstände vorliegen und der Antragsteller besondere Verfahren, insbesondere in Bezug auf die Sicherheit des Arzneimittels und die Unterrichtung der zuständigen Behörden über alle Zwischenfälle im Zusammenhang mit seiner Verwendung, einführt. Nur in objektiven und überprüfbaren Ausnahmefällen kann eine Genehmigung erteilt werden.
Bedingte Marktzulassung (Abschnitt 1.5.5)
Der erwähnte Abschnitt gilt für das zentralisierte Verfahren. Die Verweise für diesen Abschnitt sind Artikel 14 Absatz 7 der Verordnung (EG) Nr. 726/2004 und "Leitfaden zur wissenschaftlichen Anwendung und zu den praktischen Modalitäten der bedingten Marktzulassung".
Umweltrisikobewertung (Abschnitt 1.6)
Gemäß Artikel 8 (ca) und (g) der Richtlinie 2001/83/EG muss der Antragsteller bei der Beantragung der Genehmigung für das Inverkehrbringen alle potenziellen Risiken des Arzneimittels für die Umwelt berücksichtigen. Die Anforderungen der Richtlinie beziehen sich auf die Verwendung, Lagerung und Entsorgung von Arzneimitteln und gelten nicht für die Synthese oder Herstellung des Produkts. Anträge auf Genehmigung für das Inverkehrbringen von Arzneimitteln, die keine GVO enthalten (Non-GMO Abschnitt 1.6.1) und solche, die GVO enthalten (GMO 1.6.2), sollten in Modul 1 des Zulassungsdossiers einen Hinweis auf mögliche Risiken enthalten.
Weitere Informationen finden Sie unter "Leitfaden für die Umweltverträglichkeitsprüfung von Humanarzneimitteln"
Informationen zum Alleinvertrieb für seltene Leiden (Abschnitt 1.7)
Dieser Abschnitt gilt nur für Arzneimittel für seltene Leiden. Die erforderlichen Informationen über Einzelheiten und Verfahren finden Sie unter "Leitfaden der Europäischen Kommission zu Aspekten der Anwendung von Artikel 8 der Verordnung (EG) Nr. 141/2000: Bewertung der Ähnlichkeit und/oder klinischen Überlegenheit von Arzneimitteln für seltene Leiden bei der Beurteilung von Zulassungsanträgen und Änderungen."
Die Abschnitte 1.7.1 "Ähnlichkeit" und 1.7.2 "Marktexklusivität" sind anwendbar, wenn ein Arzneimittel für seltene Leiden für die Krankheit zugelassen wurde, die das vorgeschlagene therapeutische Anwendungsgebiet abdeckt, für das der Antrag gestellt wird, und wenn eine Marktexklusivitätsfrist in Kraft ist; der Antragsteller sollte einen kritischen Bericht vorlegen, der sich mit der möglichen Ähnlichkeit mit dem zugelassenen Arzneimittel für seltene Leiden befasst und zu dem Schluss kommt, dass eine Ähnlichkeit besteht oder nicht (1.7.1). Wird das Arzneimittel, für das die Genehmigung für das Inverkehrbringen beantragt wird, als "ähnlich" zu einem Arzneimittel für seltene Leiden angesehen, das unter die oben genannten Marktexklusivitätsbestimmungen fällt, muss der Antragsteller darüber hinaus eine der in Artikel 8 Absatz 3 Buchstaben a) bis c) der Verordnung (EG) Nr. 141/2000 vorgesehenen Ausnahmen begründen (1.7.2).
Information relating to Pharmakovigilanz (Abschnitt 1.8)
Pharmakovigilanz-System (Abschnitt 1.8.1)
Unter Pharmakovigilanz versteht man die Wissenschaft und die Tätigkeiten im Zusammenhang mit der Erkennung, Bewertung, dem Verständnis und der Vorbeugung von unerwünschten Wirkungen oder anderen arzneimittelbezogenen Problemen.
Dies gilt für den gesamten Lebenszyklus eines Arzneimittels, sowohl in der Phase vor der Zulassung als auch nach der Zulassung. Das Pharmakovigilanzsystem ist ein sehr wichtiger Abschnitt für die Beantragung der Genehmigung für das Inverkehrbringen.
Eine ausführliche Beschreibung des Pharmakovigilanzsystems muss gemäß Artikel 8 (ia) der Richtlinie 2001/83/EG vorgelegt werden. Die Beschreibung muss den Nachweis enthalten, dass der Antragsteller über eine qualifizierte, für die Pharmakovigilanz zuständige Person verfügt.
Die Beschreibung des Pharmakovigilanzsystems des Zulassungsinhabers sollte den Anforderungen und dem Format entsprechen, die in Band 9A von EudraLex beschrieben sind.
Risikomanagement-System (Abschnitt 1.8.2)
Detailed description of risk-management system must be provided according to Article 8 (ia) of Directive 2001/83/EC. The detailed description of a risk management system should be provided in the form of an EU Risk Management Plan (EU-RMP), as outlined in Volume 9A of EudraLex.
Informationen im Zusammenhang mit klinischen Prüfungen (Abschnitt 1.9)
Der Abschnitt 1.9 ist gemäß Artikel 8 (ib) der Richtlinie 2001/83/EG zu erstellen, damit gegebenenfalls ein Bericht vorgelegt werden kann, aus dem hervorgeht, dass die außerhalb der Europäischen Union durchgeführten klinischen Prüfungen den ethischen Anforderungen der Richtlinie 2001/20/EG entsprechen.
Dieser Abschnitt sollte für alle neuen Anträge (einschließlich Erweiterungsanträge) und andere relevante Zulassungsverfahren nach der Zulassung (z. B. Änderungen) angegeben werden, für die Berichte über klinische Prüfungen eingereicht wurden.
Informationen zur Pädiatrie (Abschnitt 1.10)
Gemäß den Artikeln 7, 8 und 30 der Verordnung (EG) Nr. 1901/2006 ("Kinderarzneimittelverordnung") und Artikel 23 der Verordnung (EG) Nr. 1901/2006 ("Kinderarzneimittelverordnung") ist dieser Abschnitt erforderlich:
- Für alle neuen Anträge* für ein Arzneimittel, das im EWR nicht zugelassen ist
- Für Anträge* auf neue Indikationen, neue Darreichungsformen und neue Verabreichungswege für zugelassene Arzneimittel, die entweder durch ein ergänzendes Schutzzertifikat oder durch ein Patent, das zur Erteilung eines solchen Zertifikats berechtigt, geschützt sind.
- Für Zulassungsanträge für die pädiatrische Verwendung (PUMA)
*ausgenommen Generika, Hybridarzneimittel, Biosimilars und Arzneimittel, die bereits seit langem verwendet werden, sowie traditionelle pflanzliche oder homöopathische Arzneimittel
Für Modul 1 können bereitgestellt werden:
Antworten auf Fragen in den Fällen, in denen den Antragstellern empfohlen wird, diesem Abschnitt ein Dokument beizufügen, in dem die Fragen mit der entsprechenden Textantwort für jede Frage aufgeführt sind, und wenn die Antworten auch neue oder aktualisierte Daten/Unterlagen zu den Modulen 3, 4 und/oder 5 enthalten. Solche Daten/Unterlagen sollten in den entsprechenden Abschnitten dieser Module aufgeführt werden.
Zusätzliche Daten. Dieser Abschnitt ist je nach Genehmigungsverfahren erforderlich. Zusätzliche Daten müssen möglicherweise als Teil eines nationalen, dezentralisierten oder gegenseitigen Anerkennungsantrags vorgelegt werden. Wenn sich diese Daten auf die Module 2, 3, 4 und/oder 5 beziehen, sollten die Dokumente auch in den entsprechenden Abschnitten dieser Module angegeben werden. Die spezifischen Anforderungen der Mitgliedstaaten für zusätzliche Daten finden Sie auf der Website der Europäische Kommission.
CTD-Baustein 2
Die allgemeine Einführung in das Arzneimittel erfolgt in Modul 2 des CTD-Dossiers, das für alle Regionen harmonisiert ist (Internationaler Rat für die Harmonisierung der technischen Anforderungen an Humanarzneimittel (ICH)). Dieses Modul wird durch zusammenfassende Dokumente für jedes kommende Modul vorgestellt: Qualitätsdaten, nicht-klinische und klinische Studienberichte.
CTD-Baustein 3
Module 3 was identified by ICH as the Quality Module. Thus, “Quality” became the global term for CMC (Chemie, manufacturing and controls). CMC ist nicht zu verwechseln mit den Elementen der Qualitätskontrolle, der Qualitätssicherung, den SOPs (Standard Operating Procedures), den internen Unternehmensdokumenten (Spezifikationen, Chargenprotokolle usw.).
Der Abschnitt des Moduls 3 ist ebenfalls für alle Regionen harmonisiert und enthält Informationen über chemisch-pharmazeutische und biologische Informationen für chemische Wirkstoffe und biologische Arzneimittel.
Während der Entwicklung und nach der Zulassung eines Produkts durch die Gesundheitsbehörden entwickeln und verändern sich die Aspekte der Chemie, Herstellung und Kontrolle (CMC) ständig weiter.
Pharmazeutische Qualität = CMC
CHEMIE
Entdeckung neuer chemischer Einheiten/Moleküle, Reinigung von Wirkstoffen (Arzneimitteln), analytische Prüfung von Rohstoffen und Fertigprodukten.
FERTIGUNG
Herstellung von Arzneimitteln und Arzneimitteln im Labormaßstab, Herstellung von klinischem Verbrauchsmaterial für klinische Studien, Aufstockung auf kommerzielle Chargengröße, kommerzielles Produkt.
UND KONTROLLEN
Entwicklung geeigneter Spezifikationen/Kontrollen für Arzneimittel und Arzneimittelprodukte zur Gewährleistung von Sicherheit, Wirksamkeit und Qualität.
Alle Regionen verlangen CMC/Pharmaqualitätsdossier
- Initiierung und Durchführung klinischer Studien,
- Einreichung eines neuen Zulassungsdossiers (MAA, NDA, BLA, etc.)
- Erfüllung der behördlichen Anforderungen für das Lebenszyklusmanagement und Änderungen nach der Zulassung des Produkts
Dieses Modul umfasst das gesamte Paket an Dokumenten zu Arzneimittelwirkstoffen (DS) und Arzneimittelprodukten (DP).
Medikamenten-Stoff (DS)
Der Abschnitt über den Arzneimittelwirkstoff wird durch seine Beschreibung, einschließlich der physikalischen, chemischen oder biologischen Eigenschaften, den Namen und die Anschrift des DS-Herstellers, die zulässigen Grenzwerte und die Analysemethoden zur Sicherstellung der Identität, Stärke, Qualität und Reinheit des Wirkstoffs dargestellt.
Außerdem sind Informationen zur Stabilität des Arzneimittels während der toxikologischen Studien und der vorgeschlagenen klinischen Studie enthalten.
DROGENPRODUKT (DP)
Dieser Abschnitt enthält eine Liste aller Bestandteile, die bei der Herstellung des Arzneimittels verwendet werden, einschließlich der Bestandteile, die im Arzneimittel enthalten sein sollen, und der Bestandteile, die zwar nicht enthalten sind, aber im Herstellungsprozess verwendet werden.
Nachfolgend sind die wichtigsten Informationen über das Arzneimittel aufgeführt, die in das Zulassungsdossier aufgenommen werden müssen:
- Die mengenmäßige Zusammensetzung des (Prüf- oder Handels-)Arzneimittels, einschließlich aller vertretbaren Abweichungen.
- Name und Anschrift des Herstellers des Arzneimittels.
- Beschreibung der Herstellungs- und Verpackungsverfahren.
- Die zulässigen Grenzwerte und Analysemethoden, die zur Sicherstellung der Identität, Stärke, Qualität und Reinheit des Arzneimittels verwendet werden.
CTD-Baustein 4
Berichte über nichtklinische Studien
Der relevante Abschnitt und der geeignete Ort für Daten zu einzelnen Tieren befindet sich im Studienbericht im Gemeinsamen Technischen Dokument für Anträge, die bei den Zulassungsbehörden eingereicht werden. Modul 4 ist auf der Grundlage der ICH-Prinzipien für die USA und die EU harmonisiert und enthält alle erforderlichen Abschnitte und Unterabschnitte für Studienberichte. Verweise auf die ICH-Richtlinien Common Technical Document (CTD).
CTD-Baustein 5
Berichte über klinische Studien
In Modul 5 geht es um die Struktur und den Inhalt von Berichten über klinische Studien. In diesem Teil des CTD werden Berichte über menschliche/klinische Studien, andere klinische Daten und Verweise innerhalb eines Gemeinsamen Technischen Dokuments (CTD) für die Registrierung eines pharmazeutischen Produkts für den menschlichen Gebrauch vorgestellt. Diese Elemente sollen die Erstellung und Prüfung eines Zulassungsantrags erleichtern. Modul 5 ist auf der Grundlage der ICH-Prinzipien für die USA und die EU harmonisiert und enthält alle erforderlichen Abschnitte und Unterabschnitte für Studienberichte. Verweise auf die EU-Leitlinien Common Technical Document (CTD).
Benötigen Sie Hilfe bei der Erstellung eines regulatorischen Dossiers? Ansehen und konsultieren Freiberufliche Verfasser von Rechtsvorschriften auf Kolabtree.
Zusätzliche Referenzen
- https://www.ich.org/page/ctd
- https://www.fda.gov/media/128163/download
- https://www.fda.gov/media/135573/download
- https://ec.europa.eu/health/sites/default/files/files/eudralex/vol-2/b/update_200805/ctd_05-2008_en.pdf
- https://ec.europa.eu/health/sites/default/files/files/eudralex/vol-1/dir_2001_83_consol_2012/dir_2001_83_cons_2012_en.pdf
- https://eur-lex.europa.eu/legal-content/EN/TXT/PDF/?uri=CELEX:02001L0083-20121116&from=DE
- https://eur-lex.europa.eu/legal-content/EN/TXT/PDF/?uri=CELEX:02004R0726-20130605&from=EN
- https://ec.europa.eu/health/sites/default/files/files/eudralex/vol-1/reg_2000_141_cons-2009-07/reg_2000_141_cons-2009-07_en.pdf
- https://www.ema.europa.eu/en/human-regulatory/research-development/clinical-trials-human-medicines
- https://eur-lex.europa.eu/legal-content/EN/TXT/?uri=celex%3A32006R1901
- https://clinicaltrials.gov/ct2/home
Kolabtree helps businesses worldwide hire freelance scientists and industry experts on demand. Our freelancers have helped companies publish research papers, develop products, analyze data, and more. It only takes a minute to tell us what you need done and get quotes from experts for free.


Unlock Corporate Benefits
• Secure Payment Assistance
• Onboarding Support
• Dedicated Account Manager
Sign up with your professional email to avail special advances offered against purchase orders, seamless multi-channel payments, and extended support for agreements.